










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Tumors arise when cells shake off their restraints and start to multiply out of control.
But how fast a tumor grows does not depend solely on how quickly the cancer cells can divide, a new study has found.
By examining brain tumors in mice, researchers at Washington University School of Medicine in St. Louis discovered that immune cells that should be defending the body against disease sometimes can be enticed into providing aid and comfort to tumor cells instead.
The more immune cells a tumor can recruit to its side, the faster the tumor grows, the researchers found.
The findings, published May 29 in the journal Neuro-Oncology, suggest that targeting immune system cells could potentially slow brain tumor growth in people with the genetic condition neurofibromatosis type 1 (NF1).
“It’s not just all about the tumor cell anymore,” said senior author David H. Gutmann, MD, Ph.D., the Donald O. Schnuck Family Professor of Neurology and director of the Washington University Neurofibromatosis Center.
“It’s also about what happens in the tumor environment that drives brain cancer growth.
This gives us another way to attack these tumors beyond merely killing the cancer cells—namely, interrupting the communication between tumor cells and immune system cells.”
While people with NF1 usually come to medical attention for birthmarks on their skin, they are also at increased risk of developing tumors.
One of the most common of these tumors in children is a low-grade brain tumor called an optic glioma, which affects the optic nerve that connects the brain and the eye. Some of these tumors can cause vision loss.
Unfortunately, NF1 is a notoriously variable disease.
Doctors can’t predict what kinds of tumors a person will develop, how fast these tumors will grow, or what types of medical problems the tumors will cause – all of which make it difficult for doctors to decide when a tumor needs to be treated with chemotherapy and when it is safe to simply watch and wait.
To better understand why some tumors grow faster than others, first author Xiaofan Guo, MD, a graduate student in Gutmann’s research laboratory, created five mouse strains with different genetic changes in the NF1 gene and elsewhere in the mouse’s genome.
The five strains varied widely in tumor development and growth.
Mice belonging to three of the strains grew tumors starting at about 3 months of age, with the tumors in one strain of mice growing particularly fast.
Members of the fourth strain didn’t grow tumors until they were about 6 months old, and only a quarter of mice in the fifth strain developed brain tumors on the optic nerve at all.
When the researchers isolated tumor cells from the mice and grew them in a dish, they found little difference in tumor cell growth.
The growth rates and other properties of the cancer cells were very similar, no matter which mutation the tumor cells carried.
What did correlate with overall tumor proliferation in mice was the presence of two kinds of immune cells – microglia and T cells – within the tumors.
Guo and former postdoctoral research fellow Yuan Pan, Ph.D., discovered that the tumor cells themselves were releasing immune system proteins that attracted immune cells to the tumor.
“Cells that should be part of the brain’s defense against tumors have become part of the process of making and growing a tumor,” said Gutmann, who is also a professor of genetics, of neurological surgery and of pediatrics.
The researchers now are trying to take advantage of this relationship between tumor cells and immune system cells to find new ways to treat brain tumors in people with NF1. One strategy is to slow tumor growth by preventing microglia or T cells from providing support to the cancer cells. However, a more ambitious strategy is to reprogram the T cells to no longer aid tumor cell growth.
“The idea is to use T cells as Trojan horses,” Gutmann said.
“These are experiments currently ongoing: We’re trying to change the T cells so that when they enter the brain, instead of promoting the tumor, they shut it down.”
How Do Cancer Cells Differ from Normal Cells?
In normal cells, hundreds of genes intricately control the process of cell division. Normal growth requires a balance between the activity of those genes that promote cell proliferation and those that suppress it. It also relies on the activities of genes that signal when damaged cells should undergo apoptosis.
Cells become cancerous after mutations accumulate in the various genes that control cell proliferation. According to research findings from the Cancer Genome Project, most cancer cells possess 60 or more mutations. The challenge for medical researchers is to identify which of these mutations are responsible for particular kinds of cancer. This process is akin to searching for the proverbial needle in a haystack, because many of the mutations present in these cells have little to nothing to do with cancer growth.
Different kinds of cancers have different mutational signatures. However, scientific comparison of multiple tumor types has revealed that certain genes are mutated in cancer cells more often than others. For instance, growth-promoting genes, such as the gene for the signaling protein Ras, are among those most commonly mutated in cancer cells, becoming super-active and producing cells that are too strongly stimulated by growth receptors. Some chemotherapy drugs work to counteract these mutations by blocking the action of growth-signaling proteins. The breast cancer drug Herceptin, for example, blocks overactive receptor tyrosine kinases (RTKs), and the drug Gleevec blocks a mutant signaling kinase associated with chronic myelogenous leukemia.
Other cancer-related mutations inactivate the genes that suppress cell proliferation or those that signal the need for apoptosis. These genes, known as tumor suppressor genes, normally function like brakes on proliferation, and both copies within a cell must be mutated in order for uncontrolled division to occur. For example, many cancer cells carry two mutant copies of the gene that codes for p53, a multifunctional protein that normally senses DNA damage and acts as a transcription factor for checkpoint control genes.
How Do Cancerous Changes Arise?
Gene mutations accumulate over time as a result of independent events. Consequently, the path to cancer involves multiple steps. In fact, many scientists view the progression of cancer as a microevolutionary process.
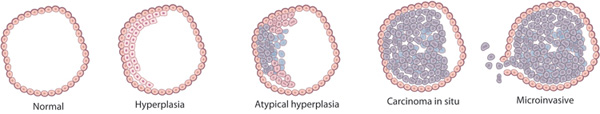

A series of mutations in colon cells can turn them into a carcinoma, a collection of cancerous cells.
© 1990 Elsevier Based on a model from Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis.
To understand what this means, consider the following: When a mutation gives a cancer cell a growth advantage, it can make more copies of itself than a normal cell can — and its offspring can outperform their noncancerous counterparts in the competition for resources. Later, a second mutation might provide the cancer cell with yet another reproductive advantage, which in turn intensifies its competitive advantage even more. And, if key checkpoints are missed or repair genes are damaged, then the rate of damage accumulation increases still further. This process continues with every new mutation that offers such benefits, and it is a driving force in the evolution of living things — not just cancer cells (Figure 1, Figure 2).
How Do Cancer Cells Spread to Other Tissues?
During the early stages of cancer, tumors are typically benign and remain confined within the normal boundaries of a tissue. As tumors grow and become malignant, however, they gain the ability to break through these boundaries and invade adjoining tissues.
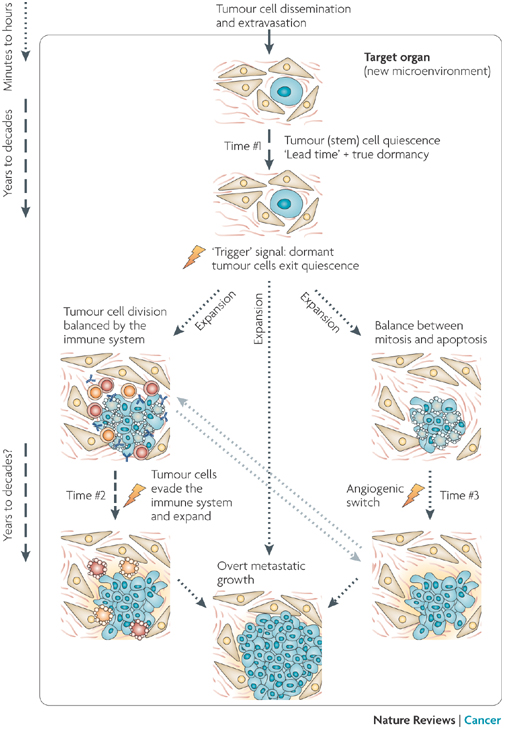
Why would metastatic cancerous cells not immediately grow new tumors? Tumor cells that survive the journey through the body will lodge in target organ tissue. This new microenvironment probably determines the fate of the disseminated tumor cells and therefore may influence the length of time cancerous cells are dormant (Time #1). If the cells do not have proper gene expression patterns, it is possible that they will be unable to grow and divide, or to transduce growth signals from the microenvironment. Consequently, they may enter a dormant state. Stress from the journey to the organ may itself trigger the quiescent state and cause growth arrest before the cell arrives at a target organ. For tumor stem cells, a quiescent state might be a natural response to a microenvironment that lacks the proper stimulating signals. Normal (nontumorous) differentiated cells can remain dormant (not grow) for years, and solitary tumor cells are found years after surgery to remove them, which suggests that a prolonged dormancy in tumor cells is plausible. Upon exit from quiescence, tumor cells can fully progress into overt lesions. It is possible that before becoming recognizable tumors, dormancy might continue (Time #2), and the immune system may also act during this time to prevent tumor expansion. After exit from quiescence or evasion of the immune system, a tumor cell mass can enter angiogenic dormancy, meaning it does not grow new blood vessels and keep growing tissue alive. It is still unclear how long (Time #3) this mechanism of angiogenic dormancy can be maintained in a genetically unstable proliferative tumor cell population. It is probable that this population would be prone to accumulating new genetic alterations that favor activation of the angiogenic switch.
© 2007 Nature Publishing Group Aguirre-Ghiso, J. A. Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer 7, 834-846 (2007) doi:10.1038/nrc2256. All rights reserved.
Invasive cancer cells often secrete proteases that enable them to degrade the extracellular matrix at a tissue’s boundary. Proteases also give cancer cells the ability to create new passageways in tissues. For example, they can break down the junctions that join cells together, thereby gaining access to new territories.
Metastasis — literally meaning “new place” — is one of the terminal stages of cancer.
In this stage, cancerous cells enter the bloodstream or the lymphatic system and travel to a new location in the body, where they begin to divide and lay the foundation for secondary tumors.
Not all cancer cells can metastasize.
In order to spread in this way, the cells must have the ability to penetrate the normal barriers of the body so that they can both enter and exit the blood or lymph vessels. Even traveling metastatic cancer cells face challenges when trying to grow in new areas (Figure 3).
More information: Xiaofan Guo et al. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia, Neuro-Oncology(2019). DOI: 10.1093/neuonc/noz080
Provided by Washington University School of Medicine in St. Louis



{kind=link}
{kind=link}