









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



More than three decades of research on Alzheimer’s disease have not produced any major treatment advances for those with the disorder, according to a UCLA expert who has studied the biochemistry of the brain and Alzheimer’s for nearly 30 years.
“Nothing has worked,” said Steven Clarke, a distinguished professor of chemistry and biochemistry. “We’re ready for new ideas.”
Now, Clarke and UCLA colleagues have reported new insights that may lead to progress in fighting the devastating disease.
Amyloid refers to the abnormal fibrous, extracellular, proteinaceous deposits found in organs and tissues.
Amyloid is insoluble and is structurally dominated by β-sheet structure. Unlike other fibrous proteins it does not commonly have a structural, supportive or motility role but is associated with the pathology seen in a range of diseases known as the amyloidoses.
These diseases include Alzheimer’s, the spongiform encephalopathies and type II diabetes, all of which are progressive disorders with associated high morbidity and mortality.
Not surprisingly, research into the physicochemical properties of amyloid and its formation is currently intensely pursued.
In this chapter we will highlight the key scientific findings and discuss how the stability of amyloid fibrils impacts on bionanotechnology.

Isolated amyloid fibrils composed of Aα chain fragment of fibrinogen (a) stained with Congo red and visualized by light microscopy and (b) between crossed polars, showing characteristic apple-green birefringence. Figure adapted from reference 83.
Scientists have known for years that amyloid fibrils – harmful, elongated, water-tight rope-like structures – form in the brains of people with Alzheimer’s, and likely hold important clues to the disease.
UCLA Professor David Eisenberg and an international team of chemists and molecular biologists reported in the journal Nature in 2005 that amyloid fibrils contain proteins that interlock like the teeth of a zipper.
The researchers also reported their hypothesis that this dry molecular zipper is in the fibrils that form in Alzheimer’s disease, as well as in Parkinson’s disease and two dozen other degenerative diseases.
Their hypothesis has been supported by recent studies.
Alzheimer’s disease, the most common cause of dementia among older adults, is an irreversible, progressive brain disorder that kills brain cells, gradually destroys memory and eventually affects thinking, behavior and the ability to carry out the daily tasks of life.
More than 5.5 million Americans, most of whom are over 65, are thought to have dementia caused by Alzheimer’s.
The UCLA team reports in the journal Nature Communications that the small protein beta amyloid, also known as a peptide, that plays an important role in Alzheimer’s has a normal version that may be less harmful than previously thought and an age-damaged version that is more harmful.
Rebeccah Warmack, who was a UCLA graduate student at the time of the study and is its lead author, discovered that a specific version of age-modified beta amyloid contains a second molecular zipper not previously known to exist.
Proteins live in water, but all the water gets pushed out as the fibril is sealed and zipped up. Warmack worked closely with UCLA graduate students David Boyer, Chih-Te Zee and Logan Richards; as well as senior research scientists Michael Sawaya and Duilio Cascio.
What goes wrong with beta amyloid, whose most common forms have 40 or 42 amino acids that are connected like a string of beads on a necklace?
The researchers report that with age, the 23rd amino acid can spontaneously form a kink, similar to one in a garden hose. This kinked form is known as isoAsp23.
The normal version does not create the stronger second molecular zipper, but the kinked form does.
“Now we know a second water-free zipper can form, and is extremely difficult to pry apart,” Warmack said. “We don’t know how to break the zipper.”
The normal form of beta amyloid has six water molecules that prevent the formation of a tight zipper, but the kink ejects these water molecules, allowing the zipper to form.
“Rebeccah has shown this kink leads to faster growth of the fibrils that have been linked to Alzheimer’s disease,” said Clarke, who has conducted research on biochemistry of the brain and Alzheimer’s disease since 1990.
“This second molecular zipper is double trouble. Once it’s zipped, it’s zipped, and once the formation of fibrils starts, it looks like you can’t stop it. The kinked form initiates a dangerous cascade of events that we believe can result in Alzheimer’s disease.”
Why does beta amyloid’s 23rd amino acid sometimes form this dangerous kink?
Clarke thinks the kinks in this amino acid form throughout our lives, but we have a protein repair enzyme that fixes them.
“As we get older, maybe the repair enzyme misses the repair once or twice,” he said.
“The repair enzyme might be 99.9% effective, but over 60 years or more, the kinks eventually build up.
If not repaired or if degraded in time, the kink can spread to virtually every neuron and can do tremendous damage.”
“The good news is that knowing what the problem is, we can think about ways to solve it,” he added. “This kinked amino acid is where we want to look.”
The research offers clues to pharmaceutical companies, which could develop ways to prevent formation of the kink or get the repair enzyme to work better; or by designing a cap that would prevent fibrils from growing.
Clarke said beta amyloid and a much larger protein tau – with more than 750 amino acids – make a devastating one-two punch that forms fibrils and spreads them to many neurons throughout the brain.
All humans have both beta amyloid and tau. Researchers say it appears that beta amyloid produces fibrils that can lead to tau aggregates, which can spread the toxicity to other brain cells.
However, exactly how beta amyloid and tau work together to kill neurons is not yet known.
In this study, Warmack produced crystals, both the normal and kinked types, in 15 of beta amyloid’s amino acids.
She used a modified type of cryo-electron microscopy to analyze the crystals. Cryo-electron microscopy, whose development won its creators the 2017 Nobel Prize in chemistry, enables scientists to see large biomolecules in extraordinary detail. Professor Tamir Gonen pioneered the modified microscopy, called microcrystal electron diffraction, which enables scientists to study biomolecules of any size.
Alzheimer’s is a disease of aggregation.
Neurons in the human brain make a protein called amyloid beta.
Such proteins on their own, called monomers of amyloid beta, perform important tasks for neurons.
But in the brains of people with Alzheimer’s disease, amyloid beta monomers have abandoned their jobs and joined together. First, they form oligomers — small clumps of up to a dozen proteins — then longer strands and finally large deposits called plaques.
For years, scientists believed that the plaques triggered the cognitive impairments characteristic of Alzheimer’s disease. But newer research implicates the smaller aggregates of amyloid beta as the toxic elements of this disease.
Now, a team led by researchers at the University of Washington has developed synthetic peptides that target and inhibit those small, toxic aggregates.
As they report in a paper published April 19 in the Proceedings of the National Academy of Sciences, their synthetic peptides — which are designed to fold into a structure known as an alpha sheet — can block amyloid beta aggregation at the early and most toxic stage when oligomers form.
The team showed that the synthetic alpha sheet’s blocking activity reduced amyloid beta-triggered toxicity in human neural cells grown in culture, and inhibited amyloid beta oligomers in two laboratory animal models for Alzheimer’s.
These findings add evidence to the growing consensus that amyloid beta oligomers — not plaques — are the toxic agents behind Alzheimer’s disease.
The results also indicate that synthetic alpha sheets could form the basis of therapeutics to clear toxic oligomers in people, according to corresponding author Valerie Daggett, a UW professor of bioengineering and faculty member in the UW Molecular Engineering & Sciences Institute.
“This is about targeting a specific structure of amyloid beta formed by the toxic oligomers,” said Daggett.
“What we’ve shown here is that we can design and build synthetic alpha sheets with complementary structures to inhibit aggregation and toxicity of amyloid beta, while leaving the biologically active monomers intact.”
Cellular proteins assume many different 3D structures, usually by first folding into certain types of basic shapes.
The alpha sheet is a nonstandard protein structure, discovered by Daggett’s group using computational simulations.
The research team has previously shown that alpha sheets are associated with aggregation of amyloid beta.
These and related findings indicate that, in nature, alpha sheets likely occur in only rare instances when proteins fold incorrectly and interact in ways that disrupt cellular function, leading to so-called “protein misfolding” diseases like Alzheimer’s.

Ball-and-stick model of the structure of AP407, one of the synthetic alpha sheet peptides designed by the research team to inhibit toxic oligomers of amyloid beta.Shea et al., PNAS, 2019
In this new paper, Daggett and her team provide evidence that amyloid beta oligomers form an alpha sheet structure as they aggregate into longer strands and plaques.
Critically, the team’s synthetic alpha sheets can actually block this aggregation by specifically binding and neutralizing the toxic oligomers.
Using both novel and conventional spectroscopic techniques, Daggett’s team observed the individual stages of development of amyloid beta clusters, from monomers to six- and 12-protein oligomers all the way up to plaques, in human neural cell lines.
The researchers confirmed that the oligomer stages were most toxic to the neurons, which agrees with clinical reports of amyloid beta plaques in the brains of people who don’t have Alzheimer’s.
“Amyloid beta definitely plays a lead role in Alzheimer’s disease, but while historically attention has been on the plaques, more and more research instead indicates that amyloid beta oligomers are the toxic agents that disrupt neurons,” said Daggett.
In addition, the researchers designed and built small, synthetic alpha sheet peptides, each made up of just 23 amino acids, the building blocks of proteins.
The synthetic peptides folded into a hairpin-like structure and are not toxic to cells.
But the synthetic alpha sheets neutralized the amyloid beta oligomers in human neural cell cultures, inhibiting further aggregation by blocking parts of the oligomers involved in the formation of larger clumps.
The peptides also protected laboratory animals from toxic oligomer damage. In brain tissue samples from mice, the team observed an up to 82% drop in amyloid beta oligomer levels after treatment with a synthetic alpha sheet peptide.
Administering a synthetic alpha sheet to living mice triggered a 40% drop in amyloid beta oligomer levels after 24 hours. In the common laboratory worm Caenorhabditis elegans, another model for Alzheimer’s disease, treatment with synthetic alpha sheets delayed the onset of amyloid beta-induced paralysis.
In addition, C. elegans worms showed signs of intestinal damage when they were fed bacteria that express amyloid beta.
That damage was inhibited when the scientists first treated the bacteria with their synthetic alpha sheets.
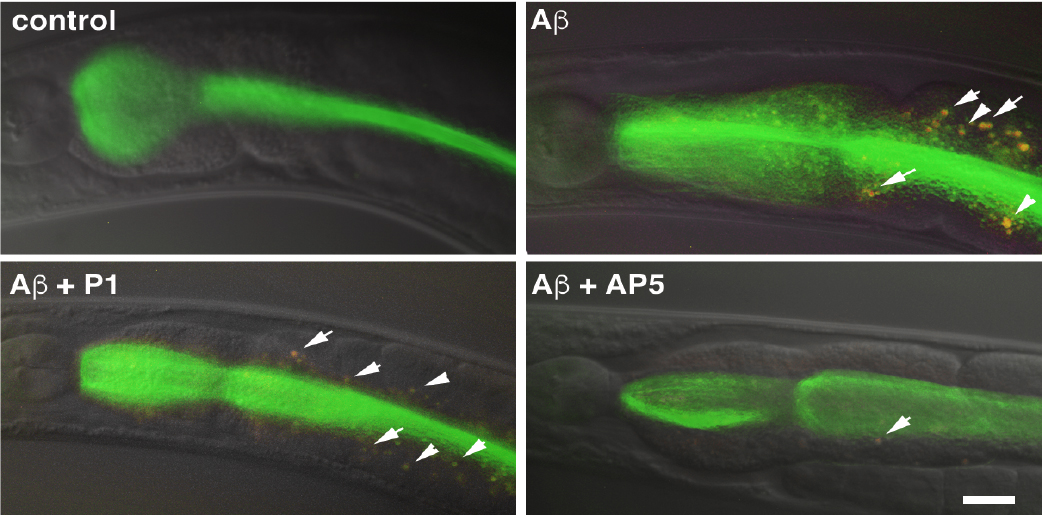
Microscopy images of the anterior intestine of four different C. elegans worms. The gastrointestinal tracts of these worms glow green because they have been fed fluorescent bacteria. Top left: A worm fed normal bacteria.
Top right: A worm fed bacteria that contain amyloid beta, which has caused intestinal damage to the worms (white arrows).
Bottom left: Intestinal damage is still present if the bacteria contain both amyloid beta and a coil protein that has no effect on aggregation. Bottom right: Intestinal damage is reduced in worms fed bacteria that express amyloid beta and AP5, a synthetic alpha sheet that blocks amyloid beta oligomers. Scale bar is 10 micrometers.Shea et al., PNAS, 2019
Daggett’s team is continuing experiments with synthetic alpha sheets to engineer compounds that are even better at clearing amyloid beta oligomers.
For the current study, the researchers also created a novel laboratory assay that uses a synthetic alpha sheet to measure levels of amyloid beta oligomers.
They believe this assay could form the basis of a clinical test to detect toxic oligomers in people before the onset of Alzheimer’s symptoms.
“What we’re really after are potential therapeutics against amyloid beta and diagnostic measures to detect toxic oligomers in people,” said Daggett. “Those are the next steps.”
Lead author is Dylan Shea, a UW doctoral student in molecular engineering. Co-authors are UW bioengineering undergraduate students Cheng-Chieh Hsu, Timothy Bi, Natasha Paranjapye and doctoral student Matthew Childers; Joshua Cochran and professor Gabriele Varani in the UW Department of Chemistry; Colson Tomberlin and associate professor Christopher Link with the University of Colorado Boulder; Libo Wang and Jeffrey Zondermanwith Redshift BioAnalytics; and Daniel Paris and executive director Mike Mullan with the Roskamp Institute. The research was funded by the National Institutes of Health, the University of Washington, the American Microscopy Society, the National Science Foundation and the Roskamp Institute.
More information: Rebeccah A. Warmack et al, Structure of amyloid-β (20-34) with Alzheimer’s-associated isomerization at Asp23 reveals a distinct protofilament interface, Nature Communications (2019). DOI: 10.1038/s41467-019-11183-z
ournal information: Nature , Nature Communications
Provided by University of California, Los Angeles



{kind=link}