










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Researchers from Duke-NUS Medical School (Duke-NUS), in collaboration with the Agency for Science, Technology and Research (A*STAR)’s Bioinformatics Institute (BII), and the University of Texas Medical Branch (UTMB), U.S., have discovered that the dengue virus changes its shape through mutations in Envelope (E) protein to evade vaccines and therapeutics.
The study also gives insights on the types of treatment strategies to use at different stages of infection. This could give rise to new approaches in vaccine development and treatment for dengue disease.
Dengue virus (DENV) infects about 400 million people annually around the world, with a high prevalence in tropical and sub-tropical regions.
The virus causes diseases ranging from mild dengue fever to severe dengue hemorrhagic fever and dengue shock syndrome.
DENV2 exists as smooth spherical surface particles while growing at the mosquito’s physiological temperature (29 degrees Celsius).
It then changes to bumpy surfaced particles at human physiological temperature (37 degrees Celsius).
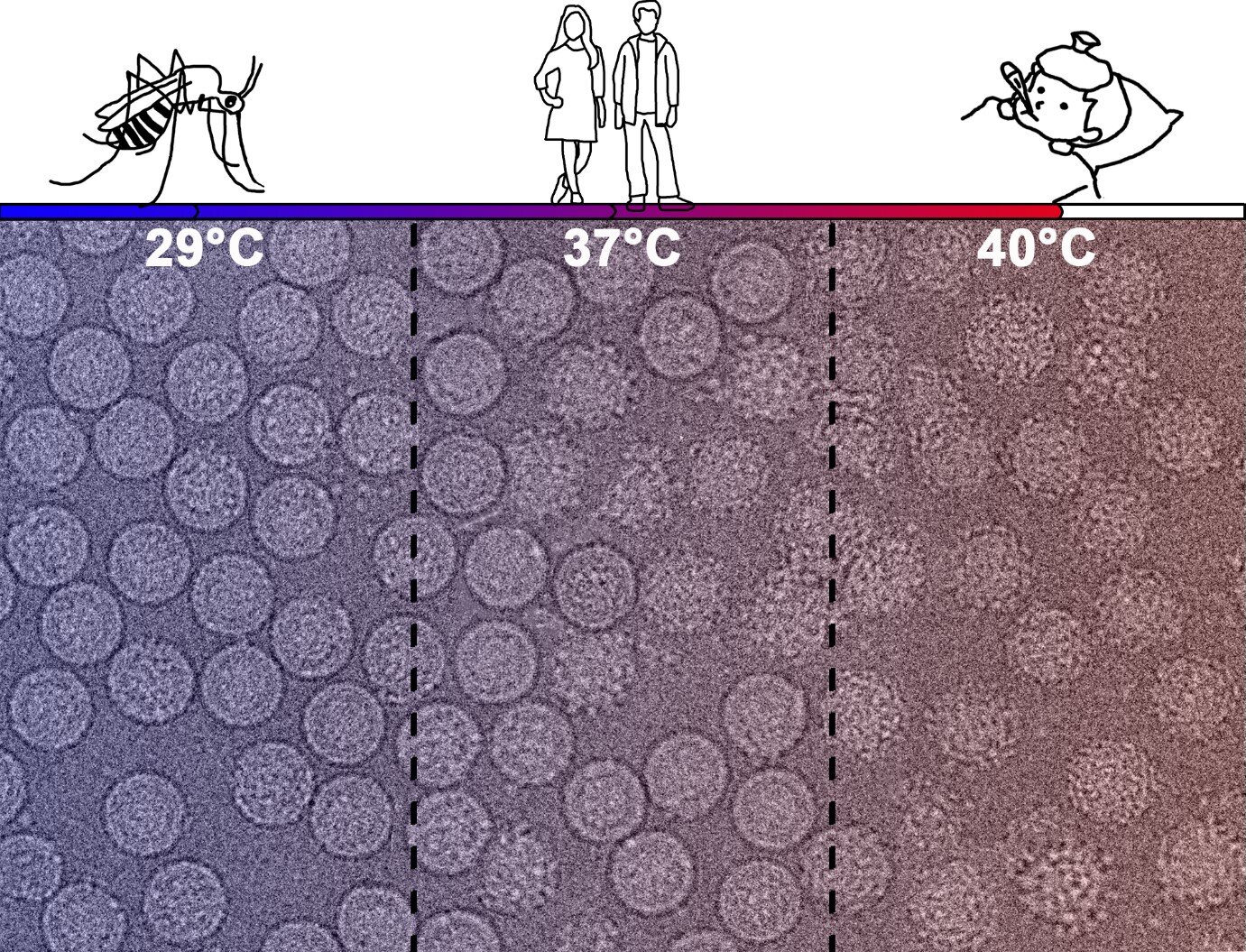
This ability to morph helps the virus to evade the immune system of the human host.
Hence, understanding the mechanism behind this is important for therapeutics and vaccine development.
“Together with Professor Pei-Yong Shi from UTMB, we found that in laboratory developed DENV2 strains, mutations in the virus’ E protein causes its transformation into bumpy particles.
These structural changes can cause vaccines and therapeutics to be ineffective against the virus,” said Ms Xin-Ni Lim, the study’s lead author who is from Duke-NUS’ Emerging Infectious Diseases (EID) Programme.
The team also tested four DENV2 strains obtained from patients.
They observed that in contrast to the laboratory adapted viruses, the majority of these clinical strains maintained smooth surface structure at 37 degrees Celsius.
However, at 40 degrees Celsius, the temperature of a fever, all virus strains took on a bumpy surface.
“Our study gives a new direction to vaccine development and treatment for dengue disease.
For prevention of disease through vaccines that are administered to the patient before dengue infection, we should use those that are effective against the smooth surface virus.
When it comes to patients displaying fever symptoms, treatment strategies effective against the bumpy surface particles should be implemented,” said Dr. Sheemei Lok, Professor, Duke-NUS’ EID and corresponding author of this study.
“This study is a first step towards gaining more insight into how DENV2 reacts and adapts to the host’s immunological defenses. We were also able to use computational modeling approaches to predict why particles from different DENV2 strains are more or less adept at morphing from the smooth to bumpy structures.
By better understanding the interactions between the virus and the host, we will be able to develop better therapies and vaccines to treat or prevent infections, and contribute to public health outcomes,” said Dr. Peter Bond, Principal Investigator from A*STAR’s BII.
The study’s findings also show that the lab adapted DENV2 may not be a good model for research, as its structure is different from the clinical strains isolated from patients. The team is planning to study the other DENV serotypes to find out if there are any other possible structural changes.
Dengue virus (DENV), a member of Flaviviridae family, contains a positive-strand (+) RNA genome and three structural proteins: envelope (E), membrane (M), and capsid (C). The E protein assembles in the virion envelope as homodimer rafts, and mediates viral entry into cells via attachment, endocytosis, and fusion with endosomal membranes—delivering the capsid into the cell cytoplasm.
The E protein folds into three major domains. Domain I (DI) is the central domain containing the protein “hinge” responsible for the low pH-catalyzed conformational change from homodimers to fusion-competent homotrimers. Domain II (DII) is the dimerization domain containing the fusion peptide at its tip.
Domain III (DIII) is an immunoglobulin-like structure suggested to mediate binding of virus to cellular receptors (Kuhn et al., 2002, Modis et al., 2003, Modis et al., 2005, Mukhopadhyay et al., 2005, Nayak et al., 2009).
DENV replication begins with the attachment of virus to cellular receptors. It is now well established that cell surface glycosaminoglycans (GAGs), such as heparan sulfate (HS), can facilitate flaviviral attachment to cultured mammalian cells (Chen et al., 1997, Germi et al., 2002, Hilgard and Stockert, 2000, Hung et al., 1999).
After solving the crystal structures of DENV2 and DENV3 E proteins, Modis et al., 2003, Modis et al., 2005 suggested that three clusters of positively charged amino acids (AAs) on E protein homodimer surface might be involved in HS binding. Cluster 1, located in DI, consists of R188, H282, K284, R286, K288, K291, and K295 (based on DENV2 E protein residue numbers) and all of these are conserved as basic residues among all 4 DENV serotypes and the Japanese encephalitis virus (JEV) complex, including West Nile virus (WNV), Murray Valley encephalitis virus (MVEV), and St. Louis encephalitis virus (SLEV). Cluster 2, located in the middle of DII near the dimer interface, includes K58, K64, K89, K122, K123, and K128 for DENV2. Unlike cluster 1, cluster 2 is less conserved among the DENV or JEV serocomplex viruses, and DENV2 contains more basic residues in this cluster than other DENV serotypes. The moderately variable cluster 3 resides (K305. K307, K310) on or near the DIII lateral ridge, including AAs important for both binding of virus-neutralizing antibody and interacting with cellular receptors (Diamond et al., 2008, Sukupolvi-Petty et al., 2007).
Because GAGs are associated with the surfaces of many cultured mammalian cells, cell-specific flaviviral attachment – if it exists – cannot be explained by interactions with only these AAs.
Cell-specific interactions that result in differential DENV tissue-tropisms in vivo probably involve other plasma membrane receptors and potentially other E protein AAs. Several candidate cell membrane proteins have been shown to bind DENV in vitro but all are still poorly characterized (Martinez-Barragan and del Angel, 2001, Ramos-Castaneda et al., 1997).
For example, it has been suggested that different cell surface proteins might be responsible for DENV binding to cultured monkey kidney cells (Vero) versus human hepatoma cells (HepG2) (Marianneau et al., 1996). Several size classes of mosquito cell (C6/36) membrane proteins appear to bind DENV, but these proteins also remain uncharacterized (Munoz et al., 1998, Salas-Benito and del Angel, 1997).
One surface protein, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), has been well-studied as a flavivirus attachment protein (Boonnak et al., 2008, Davis et al., 2006a, Davis et al., 2006b). DC-SIGN binds to virion proteins containing mannose glycans, and appears to be involved in DENV infection of primary DCs or DC-SIGN-expressing transformed cell lines—primarily through the carbohydrate moiety at the N67 glycosylation motif of the E protein (Pokidysheva et al., 2006). Since oligosaccharides on mosquito cell-derived DENVs also contain high mannose glycans, attachment to DCs via DC-SIGN may be an important first step following mosquito-bite transmission of virus to humans (Hacker et al., 2009, Hsieh and Robbins, 1984).
The region of the E protein that might bind to cell-specific receptors remains controversial, partly because the virion surface glycoprotein contains no well-defined spike structures common to other enveloped viruses (Kuhn et al., 2002, Mukhopadhyay et al., 2003, Mukhopadhyay et al., 2006, Pletnev et al., 2001, Zhang et al., 2002). DIII was first hypothesized to contain the primary receptor-binding motif because DIII-reactive neutralizing monoclonal antibodies (MAbs), and to a lesser extent DI-reactive MAbs were most effective in blocking DENV attachment to and infection of Vero cells (Crill and Roehrig, 2001). MAbs against DII epitopes had measurable, but lower, blocking activity. Several other reports also suggest that DIII binds cellular receptors (Gromowski and Barrett, 2007, Modis et al., 2003, Modis et al., 2005, Roehrig et al., 1998, Sukupolvi-Petty et al., 2007, Thullier et al., 2001, Volk et al., 2007). Several AA changes in DIII result in phenotypic changes usually associated with viral attachment, e.g., host range, tissue tropism, or virulence, and DIII also contains the most effective virus neutralization sites (Jennings et al., 1994, Modis et al., 2005, Rey et al., 1995, Roehrig, 2003, Roehrig et al., 1998). For WNV, DII also elicits effective neutralizing antibodies in humans (Vogt et al., 2009).
Hung et al., demonstrated that soluble heparin blocked binding of recombinant DIII (EIII) of DENV2 E-protein to BHK-21 cells, but not to C6/36 cells(Hung et al., 2004). They also demonstrated that a peptide representing AAs 380–389 of DIII (containing the DIII-FG loop) inhibited binding of EIII to C6/36 but not BHK21 cells.
These results suggested that DENV binding to mammalian and mosquito cells might be through different receptors. However, we have previously determined that a mutant DENV2 with a deletion of the DIII-FG loop (382VEPG385) was capable of infecting C6/36 cells, suggesting the DIII-FG loop is not the only or major receptor-binding site for C6/36 cells (Erb et al., 2010). Three basic residues, K305, K307, and K310, at a lateral ridge of DIII (DIII-A sheet in the E-dimer structure) were also speculated to form a potential HS-binding motif for DENV2 (Chen et al., 1997). A neutralizing epitope has also been mapped to K307 (Lin et al., 1994), suggesting these residues might be involved in viral attachment.
In an effort to gain a better understanding of mammalian cell attachment and entry, we report here the use of site-directed mutagenesis of a DENV2 infectious cDNA clone to introduce mutations in the HS-binding clusters and the DIII lateral ridge of the E protein. We evaluated the effects of these mutations on growth in mosquito and mammalian cells, membrane fusion in mosquito cells, attachment to Vero cells and DC-SIGN-expressing Raji cells, and expression of MAb-defined epitopes. Two Vero cell lethal mutants, KK291/295EV and KKK307/307/310EEE, were unable to initiate negative (−) sense viral (v)RNA synthesis in Vero cells by 72 h post-infection (p.i.), or replicate in DC-SIGN Raji cells, even though they were C6/36 cell-fusion competent, suggesting that these mutations abrogate an early step in viral replication, prior to virus-mediated endosomal membrane fusion. Additionally, both of these mutant viruses failed to react with MAbs shown previously to block DENV attachment to Vero cells.
More information: Xin-Ni Lim et al. Molecular basis of dengue virus serotype 2 morphological switch from 29°C to 37°C, PLOS Pathogens (2019). DOI: 10.1371/journal.ppat.1007996
Journal information: PLoS Pathogens
Provided by Duke-NUS Medical School



{kind=link}