









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Scientists at UCL have developed a method to reactivate ‘tumour suppressor’ genes, which are switched off by cancer cells – a finding which could lead to new targeted biotherapies for cancer.
In the study, published in Nature Structural and Molecular Biology, researchers at UCL Cancer Institute and the Cancer Research UK UCL Centre aimed to identify ways to block the function of a regulatory protein called PRC2 (Polycomb repressive complex 2).
Healthy cells use PRC2 to silence genes whose instructions should only be read by other cell types.
Cancer cells ‘hijack’ this silencing function of PRC2 to switch off ‘tumour suppressor’ genes.
When active, these genes stop cells from dividing, so if PRC2 could be removed from these genes it could halt tumour growth.
Using healthy cells grown in the lab, the researchers found that PRC2 also binds to RNA, the information molecule that is produced by active genes.
When PRC2 binds RNA, it can no longer bind and silence the gene.
Using cancer cells in the lab, the researchers used a protein called dCas9 (Cas9 endonuclease dead) to attach RNA to silenced tumour suppressor genes.
The RNA specifically removed PRC2 from the tumour suppressor gene to which it was attached, reactivating the gene and stopping the cancer cells from dividing.
Lead author, Professor Richard Jenner (UCL Cancer Institute), said: “Our discovery provides a way to precisely reverse cancerous gene silencing events, while leaving other genes switched off as they are supposed to be.
“The next steps are to test which cancer types this could be applied to and to develop a method that could be used to deliver the RNA and gene targeting agent to cancer cells in patients.”
Epigenetic events are key mechanisms in the regulation of cell fate and cell identity.
DNA methylation, miRNA-associated gene repression, and histone modifications have been implicated in numerous diseases, including cancers and represent new therapeutic targets [1].
Gene expression regulation through polycomb-induced histone modifications is a well-studied mechanism.
The polycomb repressive complex 2 (PRC2) contains three core subunits: EED (embryonic ectoderm development), SUZ12 (suppressor of zeste 12 homolog), and EZH2 (enhancer of zeste homolog 2). PRC2 represses gene transcription through tri-methylation of lysine 27 of histone 3 (H3K27me3) by its catalytic subunit EZH2.
EZH2 deregulation has been described in many cancer types, including hematological malignancies.
Its overexpression or gain of function mutations lead to abnormal H3K27me3 accumulation, repressing tumor suppressor genes, such as cell cycle inhibitors, apoptotic activators, and senescence and differentiation factors [1].
Multiple myeloma (MM) is a neoplasia characterized by the accumulation of clonal plasma cells within the bone marrow.
Recent advances in treatment have led to an overall survival of intensively treated patients of 6–7 years and an event-free survival of 3–4 years [2].
However, patients invariably relapse after multiple lines of treatment, with shortened intervals between relapses, and finally become resistant to all treatments, resulting in loss of clinical control over the disease.
MM is a genetically and clinically heterogeneous disease.
Genome sequencing studies have revealed considerable heterogeneity and genomic instability, a complex mutational landscape and a branching pattern of clonal evolution [3, 4].
Epigenetic marks such as DNA methylation or histone posttranslational modifications are also involved in MM pathophysiology and drug resistance [5, 6].
Global gene expression profiling indicated that, while EZH2 is upregulated, its target genes are downregulated in myeloma cells compared with normal plasma cells [7].
In human MM cell lines (HMCL), EZH2 expression has been correlated with increased proliferation and an independence on growth factors [8]. Inhibition of EZH2 expression and activity is associated with HMCL growth inhibition [9, 10] and decreased tumor load in a mouse model of MM [7, 11].
One study shows that this effect is related to epithelial tumor suppressor gene upregulation [11]. However, the use of specific EZH2 inhibitors demonstrated that MM proliferation inhibition is time dependent and cell line specific, indicating that EZH2 does not play a universal and monotonous role in promoting MM [11].
Furthermore, the first genome-wide profiling of H3K27me3 and H3K4me3 in MM patient samples was recently published, showing a unique epigenetic profile of primary MM cells compared to normal bone marrow plasma cells [10].
EZH2 inhibition was associated with upregulation of microRNAs with potential tumor suppressor functions [12]. More recently, EZH2 overexpression was reported to be associated with poor outcome and dysregulation of proliferation [13].
These data underscore an oncogenic role of EZH2 in MM. EZH2 inhibitors are currently in phase 2 clinical development in relapsed or refractory non-Hodgkin lymphoma (NHL) and biomarkers are needed for patient selection since neither EZH2 mutations nor H3K27me3 levels are sufficient to predict NHL cell response to EZH2 inhibitors [1, 14].
Here, we identified that PRC2 core genes are overexpressed in MM cells in association with proliferation activation. Treatment of MM cells with EPZ-6438, a specific small molecule inhibitor of EZH2 methyltransferase activity, results in growth inhibition due to cell cycle arrest and apoptosis.
Resistance to EZH2 inhibitor is mediated by DNA methylation of PRC2 target genes. We also observed a synergy between EPZ-6438 and lenalidomide, a conventional drug used for MM treatment. More interestingly, pretreatment of myeloma cells with EPZ-6438 significantly re-sensitizes drug-resistant MM cells to lenalidomide. EPZ-6438/lenalidomide combination induced a significant transcriptional reprogramming of MM cells targeting major B and plasma cell transcription factors in association with MYC repression. RNA sequencing combined with H3K27me3 ChIP analyses allowed us to build an EZ GEP-based score that is able to predict HMCL and primary MM cell sensitivity to EZH2 inhibitors.
PRC2 complex is overexpressed in malignant plasma cells in association with cell cycle deregulation
Using Affymetrix microarrays, we analyzed the expression of PRC2 core genes EZH2, SUZ12, and EED in normal bone marrow plasma cells (BMPCs, n = 5), primary myeloma cells from patients (MMCs, n = 206), and human myeloma cell lines (HMCLs, n = 26). PRC2 core genes are significantly overexpressed in MM cells (Fig. 1a) and EZH2 expression is significantly correlated with SUZ12 and EED expression (Additional file 1: Figure S1). At the opposite, PRC1 core genes were significantly downregulated in MM cells compared to normal plasma cells underlining polycomb complex deregulation in MM (Additional file 1: Figure S2).
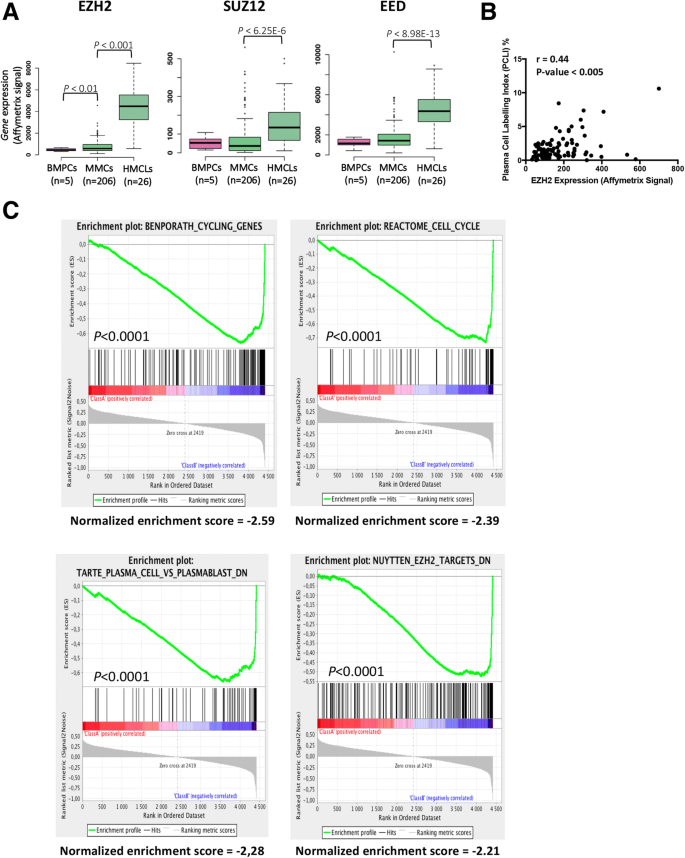
Investigating the prognostic value of EZH2, SUZ12, and EED expression in independent cohorts of previously untreated MM patients, using Maxstat R algorithm [31], only EZH2 expression was found to be associated with MM patient’s outcome as recently reported [13] (Additional file 1: Figure S3). EZH2 is significantly overexpressed in patients with del17p and 1q21 gain (Additional file 1: Figure S4A and B). EZH2 expression is significantly correlated with MMC plasma cell labeling index (PCLI) in a cohort of 101 newly diagnosed patients (P < 0.005; Fig. 1b) underlining a link between PRC2 expression and deregulation of cell cycle in MM cells. Furthermore, GSEA analysis of patients with high EZH2 expression identified a significant enrichment for genes involved in cell cycle, upregulated in proliferating plasmablasts compared to mature BMPCs, and EZH2 targets (P < 0.0001) (Fig. 1c).
Altogether, these data underline that PRC2 complex is overexpressed in MMCs in association with a proliferative plasmablastic gene signature and a poor prognosis.
To investigate the therapeutic interest of PRC2 deregulation to target MM cells, XG1, XG12, XG19, LP1, XG25, and XG7 HMCLs were treated with clinically relevant doses of EZH2 inhibitor EPZ-6438 (370 nM and 1 μM) [14, 32]. EPZ-6438 treatment induced a significant decrease of global H3K27me3 in all the HMCLs tested (P < 0.01) (Additional file 1: Figures S5 and S6) and inhibited MM cell growth together with proliferation inhibition and apoptosis induction in 3 out of the 6 HMCLs tested (Fig. 2a and Additional file 1: Figure S7). The inhibitory effect appeared at day 6, suggesting that it is mediated by epigenetic reprogramming (Fig. 2a). EZH2 inhibitor induced apoptosis was partially rescued by the QVD pan-caspase inhibitor, suggesting a caspase-dependent mechanism (Additional file 1: Figure S7). LP1 and XG7 were more resistant to EZH2 inhibitor whereas XG25 HMCL was completely resistant. Primary MM cells co-cultured with their bone marrow microenvironment and recombinant IL-6 were also treated with EPZ-6438 as previously described [6]. EZH2 targeting significantly reduced the median number of viable myeloma cells by 35% (P = 0.004) in 9/17 patients whereas MM cells of 8 patients were not significantly affected by the EZH2 inhibitor (Fig. 2b and Additional file 1: Figure S8). As described in HMCLs, EPZ-6438 induced a significant global H3K27me3 decrease in all the patients (Fig. 2c). The effect of EZH2 inhibitor was not correlated with EZH2 expression, H3K27me3 and levels in light of the 6 HMCLs and 17 primary MM samples tested. Moreover, UTX/JMJD3 demethylases mutation status did not seem to affect EPZ-6438 efficiency in the tested samples (Additional file 1: Figure S9 and Additional file 2: Table S1).
We therefore conclude that MM cell growth could be affected by PRC2 targeted therapy which presents a therapeutic interest in a subgroup of MM patients.
More information: G-tract RNA removes Polycomb repressive complex 2 from genes, Nature Structural and Molecular Biology (2019). DOI: 10.1038/s41594-019-0293-z , https://nature.com/articles/s41594-019-0293-z
Journal information: Nature Structural and Molecular Biology
Provided by University College London



{kind=link}