










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



No matter the pathogen – virus, bacterium or fungus – many “pick a specific type of lock” on the surface of cells, which allows the microbe to break and enter into the inner sanctum of the host’s genome.
In a sweeping analysis on how microbes hack into cells and commandeer them, Australian microbiologists say they are also uncovering mechanisms in common between microbes and cancer cells.
Their research suggests that in the not-too-distant future it may be possible to cure infectious diseases with repurposed cancer drugs, medications that can function across a broad range of pathogens, eliminating the need for antibiotics, antivirals and antifungals.
An ultimate hope, these scientists say, is ushering in the era of anti-infectives, drugs that block a vulnerable port of entry for infectious agents.
Writing in the journal Science Signaling, Dr. Gholamreza Haqshenas of Monash Biomedicine Discovery Institute in Australia, and Dr. Christian Doerig, also of Monash, as well as RMIT University in Australia, say a radically diverse group of pathogens have surprisingly evolved to subvert cells in a sly way.
While there are numerous pathways to infection, many microbes enter their hosts by hijacking signaling proteins, a finding that has become increasingly clear in recent years, Haqshenas and Doerig say.
Signaling proteins are molecules that are responsible for sending messages within and between cells.
These proteins have segments above and below the cell surface, which are critical to cellular function.
So it should come as no surprise, the Australian team says, that microbes responsible for some of the most serious infections have targeted a superfamily of signaling molecules—receptor tyrosine kinases—as their route into the host’s cellular domain.
Haqshenas said a vast range of pathogens have evolved mechanisms to hack into cells by targeting receptor tyrosine kinases, or RTKs. These microbes can bind to the receptor and like a safe cracker, “unlock” the cell.
“To name some important ones: Hepatitis C and influenza viruses,” Haqshenas told Medical Xpress.
“Among bacteria, Salmonella and Listeria monocytogenes; and among fungi, Candida albicans.”
When a bacterium, such as Chlamydia pneumoniae binds to a receptor tyrosine kinase protein on a cell’s surface, the pathogen not only commandeers the cell, it stimulates signaling, the messages that control the cell.
The pathogen assumes command of everything including the cell’s structure, its cytoskeleton, thereby easing its entry into the cell.
The RTK superfamily, which has about 58 members, function as the cell receptors for numerous biological growth factors, such as epidermal growth factor, platelet-derived growth factor and vascular endothelial growth factor, to name a few.
These factors are like keys that once bound to the receptor can unlock it and enter the cell. An overabundance of RTKs occurs in some cancers, which can lead to cancer progression. RTK blockers, medications referred to as small-molecule drugs, have been developed to treat a form of leukemia and a rare intestinal cancer, which are marked by excessive RTKs. Gleevec was the first drug developed in this class and was approved by the U.S. Food and Drug Administration nearly two decades ago.
Although Haqshenas and Doerig report that a veritable rogue’s gallery of viruses can commandeer receptor tyrosine kinases as an entryway into cells, Doerig underscored “there are other mechanisms of entry for some bacteria.”
Pathogens that use the RTK route to infection have evolved specialized mechanisms that have allowed them to hack their way into host cells via this passageway.
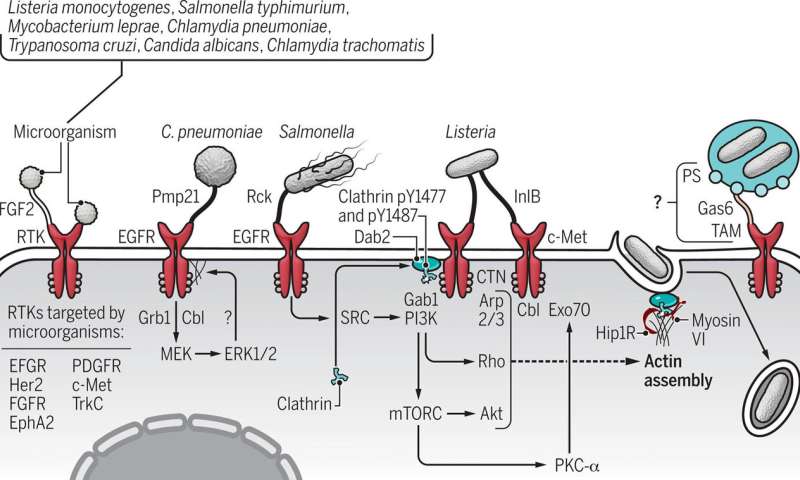
For example, Chikungunya, Ebola, Eastern equine encephalitis and Japanese encephalitis viruses enter cells via RTKs.
In addition, cytomegalovirus, dengue, herpes simplex virus 1, human papillomavirus, Lassa, Marburg, respiratory syncytial, yellow fever, West Nile and Zika viruses break and enter through the receptor tyrosine kinase pathway.
The list is longer still, according to Haqshenas and Doerig, who say it also includes Kaposi’s sarcoma-associated herpesvirus, lymphocytic choriomeningitis, Rose River and vaccinia viruses, pathogens that also preferentially hack their way into host cells by unlocking RTKs.
“The commonality of relying on the same RTKs suggests that selective inhibitors against specific RTKs may have broad spectrum anti-infective properties. Definitely worth further investigation,” Doerig said.
Haqshenas likewise sees their new analysis as a springboard to further research that explores the possibility of using existing drugs in new ways against infectious agents.
“Our review highlights the importance of RTKs in the biology of many pathogens of importance to public health,” Haqshenas said. “Currently, RTKs are common targets of anti-cancer drugs. The review highlights that FDA-approved anti-cancer drugs can be repurposed for the treatment of infections caused by a wide range of pathogens that must enter host cells to complete their life cycle.
“This approach will significantly reduce the cost of anti-infective drug development, and, because a cellular protein is targeted, it reduces the chance of drug resistance,” Haqshenas said.
Bacteria and fungi are highly efficient in acquiring antimicrobial resistance encoded by genomic changes ranging in scale from point mutations, through the assembly of preexisting genetic elements, to the horizontal import of genes from the environment (Kung et al., 2010; Cowen et al., 2015; Yelin and Kishony, 2018). Compounding the problem of antimicrobial resistance is the immediate threat of a reduction in the discovery and development of new antibiotics, the dangers of which have recently been made clear by the World Health Organization (WHO) (Tacconelli et al., 2018) and other European institutions (O’Neill, 2016; Årdal et al., 2018). Consequently, a perfect storm is converging with regard to these infections: increasing antimicrobial resistance with a decreased new drug development (O’Neill, 2016). This context is likely the best example of the purported “Post-Antibiotic Era,” with relevance even in non-specialized media (Bagley and Outterson, 2017). It is clear that effective solutions are urgently needed as stressed by various institutions.
New policies and actions are necessary to avoid the figures predicted for 2050 that attribute ten million deaths worldwide to antimicrobial resistance (O’Neill, 2016). Such efforts might include: a massive global public awareness campaign; improvements in hygiene and prevention of the spread of infection; increase global surveillance of drug resistance and the appropriate antimicrobial consumption in humans and animals; the promotion of novel rapid diagnostics to curtail the unnecessary use of antimicrobial agents; and the promotion, development, and use of vaccines and other alternatives to both prevent and treat bacterial infections (O’Neill, 2016). Therefore, the development of new antimicrobial therapeutic strategies for use alone or together with one of the scarce but clinically relevant antibiotics has become exigent. In this environment, “repurposing” (defined as investigating new uses for existing drugs) has gained renewed interest, as reflected by several recent studies (Fischbach and Walsh, 2009; Brown, 2015; Rampioni et al., 2017). The combination of these existing drugs with antimicrobial agents currently in clinical use is also under consideration.
A literature review was conducted to search for potential non-antimicrobial candidate drugs that are not intentionally used as antimicrobial agents but have one or more antimicrobial properties. A variety of drug families have been considered including: anthelmintics (Lim et al., 2013; Rajamuthiah et al., 2015; Gooyit and Janda, 2016; Joffe et al., 2017); anticancer drugs (Ueda et al., 2009; Butts et al., 2014; Yeo et al., 2018); anti-inflammatory/immunomodulatory drugs (Artini et al., 2014; Thangamani et al., 2015b; Ogundeji et al., 2016); antipsychotic and antidepressant drugs (Lieberman and Higgins, 2009; Andersson et al., 2016; Holbrook et al., 2017); statins (Parihar et al., 2014; Thangamani et al., 2015a; Ribeiro et al., 2017); and iron-storage drugs (Gi et al., 2014). While these drugs are approved for their clinical indications, promising antibacterial and antifungal activities have been reported in preclinical and clinical studies. It is noteworthy that repurposing drugs is a very promising approach with several advantages. As drugs approved by the Federal Drug Administration (FDA), information about their pharmacological characteristics (both safety and pharmacokinetic) in preclinical and clinical trials is widely available. Therefore, the time and economic costs associated with the repurposing of these drugs for other therapeutic applications such as the treatment of bacterial and fungal infections will be minimized.
In this review, we focus on the current state of knowledge regarding the repurposing of drugs in terms of their modes of action, antimicrobial efficacy and breadth of spectrum against bacteria and fungi, as well as the advances to-date in their development as antimicrobial agents for clinical use. To this end, we introduce in Pubmed database different key words such as repurposing drugs, antibacterial and/or antifungal in order to find published literature about the repurposing drugs for treatment of bacterial and fungal infections.Go to:
Potential Drugs for Repurposing Against Infectious Agents
The antibacterial and antifungal activities of repurposing drugs and their modes of action are summarized in Table 1 and Figure 1.
Table 1
Direct antibacterial and antifungal activity of repurposed drugs.
| Drugs | Clinical indication | Target bacteria | Mechanisms of action | Reference |
|---|---|---|---|---|
| Niclosamide∗ | Helminthiasis | P. aeruginosa | Inhibition of quorum sensing and various virulence genes, and reduction of elastase and pyocyanin levels | Imperi et al., 2013b |
| Oxyclozanide | Helminthiasis | S. aureus | Bacterial membrane damage | Rajamuthiah et al., 2015 |
| Mebendazole | Helminthiasis | C. neoformans | Morphological alterations by reducing capsular dimensions | Joffe et al., 2017 |
| Quinacrine | Helminthiasis | C. albicans | Inhibition of filamentation | Kulkarny et al., 2014 |
| Floxuridine | Colorectal cancer | S. aureus | Inhibition of the SaeRS two-component system, and inhibition of the transcription of other virulence regulatory systems | Yeo et al., 2018 |
| Streptozotocin | Pancreatic islet cell cancer | S. aureus | Inhibition of the SaeRS two-component system, and inhibition of the transcription of other virulence regulatory systems | Yeo et al., 2018 |
| Toremifene | Breast cancer | S. mutans and P. gingivalis | Membrane permeabilization and damage | Gerits et al., 2017 |
| C. neoformans | Binding to the two essential EF-hand proteins calmodulin 1 (Cam1) and calmodulin-like protein (Cml) and prevention of Cam1 from binding to its well-characterized substrate calcineurin | Butts et al., 2014 | ||
| Tamoxifen | Breast cancer | C. neoformans | Binding to the two essential EF-hand proteins calmodulin (Cam1) and calmodulin-like protein (Cml) and prevention of Cam1 from binding to its well-characterized substrate calcineurin | Butts et al., 2014 |
| Raloxifene | Breast cancer | P. aeruginosa | Binding to PhzB2 which is involved in the production of pyocyanin, a pigment related with the virulence of P. aeruginosa | Ho Sui et al., 2012 |
| Clomiphene | Fertility | S. aureus | Inhibition of undecaprenyl diphosphate synthase involved in the synthesis of teichoic acid wall | Farha et al., 2015 |
| Finasteride | Benign prostatic hyperplasia | C. albicans | Inhibition of filamentation | Routh et al., 2013 |
| 5-fluorouracil | Solid tumors | P. aeruginosa | Inhibition of biofilm formation and quorum sensing | Ueda et al., 2009 |
| Doxorubicin | Bladder, breast, stomach, lung, ovarian, and thyroid cancers | C. albicans | Inhibition of filamentation | Chavez-Dozal et al., 2014 |
| Daunorubicin | Acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, and Kaposi’s sarcoma | C. albicans | Inhibition of filamentation | Chavez-Dozal et al., 2014 |
| Clofoctol | Bacterial infection | P. aeruginosa | Inhibition of the pqs system, probably by targeting the transcriptional regulator PqsR | D’Angelo et al., 2018 |
| Azithromycin | Bacterial infection | P. aeruginosa | Interaction with the ribosome, resulting in direct and/or indirect repression of specific subsets of genes involved in virulence, quorum sensing, biofilm formation, | Imperi et al., 2014 |
| 5-fluorocytosine | Fungal infection | P. aeruginosa | Inhibition of the production of pyoverdine, Prlp protease, and exotoxin A by downregulation of the pvdS gene. | Imperi et al., 2013a |
| Clotrimazole and miconazole | Fungal infection | P. aeruginosa | Inhibition of the pqs activity through the possible inactivation of 2-alkyl-4-quinolones (AQ) production or reception | D’Angelo et al., 2018 |
| Gallium nitrate∗ | Lymphoma and bladder cancer | P. aeruginosa | Effects on iron metabolism | Antunes et al., 2012 |
| Celecoxib | Inflammation | S. aureus, B. anthracis, B. subtilis, and M. smegmatis | Inhibition of bacterial DNA, RNA, protein synthesis, and cell wall | Thangamani et al., 2015b |
| Diflunisal | Inflammation | S. aureus | Inhibition of ArgA, a regulator protein which inhibits alpha-type phenol soluble modulins toxins | Hendrix et al., 2016 |
| Glatiramer acetate | Inflammation | P. aeruginosa | Disruption of biofilm formation | Christiansen et al., 2017 |
| Aspirin and ibuprofen | Inflammation | C. neoformans and C. gattii | Stress induction via activation of the high-osmolarity glycerol (HOG) pathway, and activation of reactive oxygen species (ROS)-mediated membrane damage | Ogundeji et al., 2016 |
| Pimozide | Severe Tourette’s syndrome and schizophrenia | L. monocytogenes | Reduction of L. monocytogenes internalization by phagocytic cells by decreasing vacuolar escape and diminishing cell-to-cell spread | Lieberman and Higgins, 2009 |
| Azathioprine | Crohn’s disease | P. aeruginosa and E. coli | Inhibition of WspR. WspR is a diguanylate cyclase involved in the regulation of a signal molecule called cyclic-di-GMP (c-di-GMP) known as a regulated of the bacterial biofilm formation | Antoniani et al., 2013 |
| Simvastatin | Hypercholesterolemia | M. tuberculosis | Reduction of cholesterol within phagosomal membrane | Parihar et al., 2014 |
| Atorvastatin | Hypercholesterolemia | C. gattii | Reduction of the ergosterol content in the cell membrane and alteration of the properties of the polysaccharide capsule; increase in the production of ROS by macrophages; and reduction of yeast phagocytosis and the intracellular proliferation rate | Ribeiro et al., 2017 |
| Ebselen∗ | Bipolar disorder and ischemic stroke | S. aureus, C. difficile | Reduction of biofilm formation and targeting of the glucosyltransferase domain toxins | Gi et al., 2014; Peng et al., 2018 |
| Pentetic acid | Hypocalcemia | P. aeruginosa | Reduction of biofilm formation and inhibition of elastase | Gi et al., 2014 |
| Auranofin | Rheumatoid arthritis | S. aureus | DNA inhibition and protein synthesis, and downregulation of toxin production | Thangamani et al., 2016a |
Open in a separate window∗ Some of the drugs listed in this table displayed multiple effects against different pathogens, and that only the findings of an exemplifying study are reported in the table. Other studies on the same drugs are discussed in the text.

Antibacterial and antifungal mechanisms of action of repurposed drugs.
Anthelmintic Drugs Repurposed Against Bacteria and Fungi
Anthelmintic drugs constitute a large class of medications used for the treatment of helminthiasis. Their activities aside from their use against parasitic infections are being investigated in other areas such as oncology (Dogra et al., 2018; Wang et al., 2018). The activity of these drugs against Gram-positive and Gram-negative bacteria, and fungi has been reported. The salicylanilide family contains a number of the anthelmintic drugs approved for the treatment of parasitic infections. The most widely used members of this family include niclosamide for humans (Chen et al., 2018) and oxyclozanide, rafoxanide, and closantel for animals (Martin, 1997).
The mode of action of salicylanilides is not completely understood, but they are thought to act as uncouplers of oxidative phosphorylation, thereby impairing the motility of parasites. Rajamuthiah et al. (2015) described the efficacy of niclosamide and oxyclozanide against methicillin-, vancomycin-, linezolid-, or daptomycin-resistant Staphylococcus aureus isolates. They reported that niclosamide presented bacteriostatic activity whereas oxyclozanide exhibited antibacterial action, likely due to damage in the bacterial membrane. Together with niclosamide and oxyclozanide, other members of the salicylanilides family such as rafoxanide and closantel have presented greater bactericidal activity against the logarithmic and stationary phases of Clostridium difficile than vancomycin (Gooyit and Janda, 2016). Avermectins, a broad-spectrum class of anthelmintic drugs which include ivermectin, selamectin, and moxidectin, demonstrated efficacy in vitro against Mycobacterium tuberculosis and Mycobacterium ulcerans with minimum inhibitory concentration (MIC) values ranging from 1 to 8 mg/L and 4 to 8 mg/L, respectively (Lim et al., 2013; Omansen et al., 2015). Moreover, ivermectin showed efficacy against S. aureus clinical isolates including methicillin-resistant strains (MRSA) (Ashraf et al., 2018). In vivo, ivermectin improves LPS-induced survival in mice by reducing serum and murine macrophage levels of TNF-α, IL-1β, and IL-6 and blocking the NF-κB pathway (Zhang et al., 2008).
In Gram-negative bacteria, only niclosamide exhibited antibacterial activity. This drug showed an anti-virulent effect against Pseudomonas aeruginosa via the inhibition of quorum sensing and virulence genes, reducing elastase and pyocyanin levels (Imperi et al., 2013b). In Acinetobacter baumannii and Klebsiella pneumoniae, niclosamide is able to increase the proportion of negative charges on their cell walls, and to potentiate the activity of colistin against colistin-resistant A. baumannii and K. pneumoniae (Ayerbe-Algaba et al., 2018). Recently, the effectiveness of niclosamide against Helicobacter pylori has been described, showing an MIC of 0.25 mg/L against the ATCC 49503 strain (Tharmalingam et al., 2018). Furthermore, niclosamide has demonstrated an immunomodulatory role by decreasing the secretion of IL-8 in a gastric cancer cell line after H. pylori infection (Tharmalingam et al., 2018). Niclosamide also showed therapeutic efficacy in an experimental infection model of Galleria mellonella larvae infected with P. aeruginosa and H. pylori (Imperi et al., 2013b; Tharmalingam et al., 2018). The formulation of niclosamide under nanosuspension showed lower toxicity in a rat lung infection model involving P. aeruginosa; the results of this study are potentially favorable for the further study of this formulation (Costabile et al., 2015).
In the case of fungi, mebendazole inhibited the growth of Cryptococcus neoformans and Cryptococcus gattii and affected the formation of biofilm by C. neoformans (Joffe et al., 2017). The combination of mebendazole with amphotericin B increased the fungicidal activity of amphotericin B against C. neoformans twofold (Joffe et al., 2017). Moreover, quinacrine, in monotherapy, has been shown in vitro to be effective for the prevention and treatment of Candida albicans biofilms, accumulating in vacuoles and causing defects in endocytosis (Kulkarny et al., 2014). In combination with caspofungin or amphotericin B, quinacrine has demonstrated synergy against C. albicans (Kulkarny et al., 2014).
These studies highlight the potential use of the anthelmintic drugs as antimicrobial agents as monotherapy for infections caused by Gram-positive and Gram-negative bacteria and fungi; although in vivo studies in vertebrate experimental models should be conducted.
Anticancer Drugs Repurposed Against Bacteria and Fungi
The antibacterial activity of anticancer drugs has also been reported (Soo et al., 2017). Most of them act against Gram-positive pathogens.
The FDA-approved anticancer drugs floxuridine (mostly used in colorectal cancer) and streptozotocin (used for pancreatic islet cell cancer) have exhibited activity against S. aureus by inhibiting the SaeRS two-component system (TCS) (Yeo et al., 2018). SaeRS TCS is an important transcriptional regulator of different virulence factors of S. aureus including adhesins, toxins, and enzymes (Yeo et al., 2018). Floxuridine showed direct antibacterial activity by inhibiting the growth of S. aureus USA300 at a concentration of 0.0625 mg/L in vitro and increasing the survival of mice by 60% in a murine model of blood infection in vivo (Yeo et al., 2018). On the other hand, streptozotocin did not affect staphylococcal growth in vitro but reduced the mortality of mice to 10% in vivo (Yeo et al., 2018). Both drugs not only cause significant changes in the transcription of S. aureus genes, but also inhibit the transcription of other virulence regulatory systems of S. aureus (Yeo et al., 2018).
Another group of anticancer drugs developed to combat breast cancer is the selective estrogen receptor modulators (SERMs). Tamoxifen has been reported to exhibit activity against S. aureus (Corriden et al., 2015) and its analog toremifene showed efficacy against oral infection caused by Streptococcus mutans (Gerits et al., 2017). Toremifene also has been shown to reduce biofilm formation by S. mutans due to a possible increase in membrane permeabilization and therefore, membrane damage (Gerits et al., 2017). Clomiphene, another SERM in preclinical development for the treatment of fertility, has demonstrated efficacy against S. aureus and Bacillus subtilis in vitro, with an MIC value of 8 mg/L, and incubation of B. subtilis with this concentration of clomiphene changed its morphology (Farha et al., 2015). The mode of action of clomiphene is through the inhibition of undecaprenyl diphosphate synthase (UPPS), an enzyme involved in the synthesis of the teichoic acid wall of S. aureus (Farha et al., 2015). Due to this action on the bacterial wall, clomiphene exhibits synergy with β-lactams in restoring MRSA susceptibility (Farha et al., 2015).
Other anticancer drugs were tested as adjunctive therapies against M. tuberculosis infection. One such drug, denileukin diftitox, is currently used for the treatment of cutaneous T-cell lymphoma (Gupta et al., 2017). Treatment with denileukin diftitox slightly reduced the lung bacterial count in mice aerosol-infected with M. tuberculosis (Gupta et al., 2017). The addition of this drug to standard tuberculosis treatments, composed of rifampin, isoniazid, and pyrazinamide, similarly reduced the bacterial burden (Gupta et al., 2017).
Different studies have been also performed on Gram-negative bacteria to evaluate the antibacterial effect of anticancer drugs. A potent anticancer drug indicated for the treatment of different types of solid tumors called 5-fluorouracil, as well as gallium nitrate, an anticancer drug for the treatment of lymphoma and bladder cancer, have been extensively studied (Banin et al., 2008; Bonchi et al., 2014; Minandri et al., 2014; Rangel-Vega et al., 2015). 5-fluorouracil has been used against a collection of 5,850 mutants of the P. aeruginosa PA14 strain, revealing positive activity via the regulation of a large number of genes involved in quorum sensing and biofilm formation (Ueda et al., 2009; Rangel-Vega et al., 2015). Also, gallium nitrate has demonstrated in vitro an inhibitory effect on bacterial growth in a collection of 58 multidrug-resistant (MDR) A. baumannii strains, and in P. aeruginosa at concentrations >3.13 μM (Kaneko et al., 2007; Antunes et al., 2012; Frangipani et al., 2014); although the presence of pyoverdine and proteases in human serum reduce the efficacy of gallium nitrate against P. aeruginosa by increasing its MIC (Bonchi et al., 2015). At non-bactericidal concentrations, gallium nitrate can affect the production of virulence factors of P. aeruginosa (Kaneko et al., 2007; García-Contreras et al., 2014). In G. mellonella, the administration of this drug alone or in combination with colistin, at concentrations mimicking the human therapeutic dose of gallium nitrate used for cancer patients (28 μM), significantly increased the survival of larvae after infection by A. baumannii (Antunes et al., 2012). Moreover, in murine models of acute and chronic lung infections by P. aeruginosa and A. baumannii, gallium nitrate has reduced lung injury and bacterial loads in tissues (Kaneko et al., 2007; de Léséleuc et al., 2012). Regarding SERM drugs, toremifene has shown efficacy against oral infection caused by Porphyromonas gingivalis (Gerits et al., 2017), and raloxifene attenuated in vitro and in Caenorhabditis elegans model the virulence of P. aeruginosa by binding to PhzB2 which is involved in the production of pyocyanin, a pigment related with the virulence of this pathogen (Ho Sui et al., 2012).
In the case of fungi, the activity of anticancer drugs has also been investigated. Floxuridine, at twice its half maximal inhibitory concentration (IC50) value, has exhibited fungicidal activity against Exserohilum rostratum reducing the hyphae-derived CFU (colony-forming unit) of this fungus (Sun et al., 2013). The SERM compounds such as tamoxifen and toremifene have also shown fungicidal activity against C. neoformans. They also display a number of pharmacological properties desirable for anticryptococcal drugs, including synergistic fungicidal activity with fluconazole and/or amphotericin B in vitro and in vivo, oral bioavailability, and activity within macrophages (Butts et al., 2014). They bind directly to the two essential EF-hand proteins calmodulin 1 (Cam1) and calmodulin-like protein 1 (Cml1) of C. neoformans, preventing Cam1 from binding to its well-characterized substrate calcineurin (Cna1), thereby blocking Cna1 activation (Butts et al., 2014). In whole cells, toremifene and tamoxifen are known to block the calcineurin-dependent nuclear localization of the transcription factor Crz1 (Butts et al., 2014). Together, both drugs have inhibited the growth of C. neoformans within macrophages, a niche not accessible to current antifungal drugs (Butts et al., 2014). In murine-disseminated cryptococcosis, tamoxifen in combination with fluconazole decreased the brain burden by ∼1 log10 CFU/g (Butts et al., 2014). Against C. albicans and Candida glabrata, toremifene has enhanced the antibiofilm activity of amphotericin B and caspofungin [fractional inhibitory concentration index (FICI) < 0.5 both in vitro and in vivo worm infection models (Delattin et al., 2014)].
Other anticancer drug such as finasteride, a 5-α-reductase inhibitor commonly used for the treatment of benign prostatic hyperplasia, was highly effective in both the prevention and destruction of C. albicans biofilm formation at doses greater than 16 and 128 mg/L, respectively (Chavez-Dozal et al., 2014). In combination with 2 mg/L fluconazole, 2 mg/L, finasteride exhibited synergistic activity in the prevention of biofilm formation by C. albicans (Chavez-Dozal et al., 2014). Similar effects were observed in the presence of doxorubicin and daunorubicin that inhibited the morphogenesis of C. albicans (Routh et al., 2013).
Anticancer drugs not only target bacteria and fungi but can also regulate the host response. Floxuridine and streptozotocin have presented a protective effect on the host by reducing S. aureus-mediated killing in human neutrophils (Yeo et al., 2018). Moreover, tamoxifen can stimulate chemotaxis, phagocytosis, and neutrophil extracellular trap (NET) formation through the modulation of the ceramide pathway upon infection with S. aureus (Corriden et al., 2015). Unlike tamoxifen, its analog raloxifene has been shown to reduce NET formation in human neutrophils, thus resulting in cell death of S. aureus (Flores et al., 2016). In addition, denileukin diftitox has been reported to bind to the IL-2 receptor in T lymphocytes, thereby introducing diphtheria toxin inside these cells to suppress them. The decrease in this type of T cell hinders the replication of M. tuberculosis (Gupta et al., 2017). It is noteworthy to mention that toxicity of anticancer drugs is important in terms of their establishment as antibacterial drugs. Tamoxifen has been used for over 30 years to treat breast cancer. The doses of tamoxifen used in animals (250 mg/kg) (Corriden et al., 2015) and in humans (20–40 mg) are generally tolerated. For clomiphene, acute oral LD50 in mice and rats were 1,700 and 5,750 mg/kg, respectively (Drug Bank, 2018). The toxic dose of clomiphene in humans is unknown but toxic effects accompanying acute overdosage were not observed (Drug Bank, 2018). In the case of gallium nitrate, the treatment of hypercalcemia was performed with continuous intravenous infusion (200 mg/m2/day during 5 days) being generally well tolerated (Warrell et al., 1988). On the other hand, higher doses (300 mg/m2/day during 5–7 days) were used in cancer and side effects such as diarrhea and renal toxicity were observed (Chitambar, 2010).
Anti-inflammatory and Immunomodulatory Drugs Repurposed Against Bacteria and Fungi
As is the case with anticancer drugs, anti-inflammatory and immunomodulatory drugs have demonstrated more antibacterial activity against Gram-positive than Gram-negative bacteria and fungi.
Celecoxib, a non-steroidal anti-inflammatory drug (NSAID), showed antibacterial activity against several pathogens including S. aureus, Bacillus anthracis, B. subtilis, and M. smegmatis (Thangamani et al., 2015b). Celecoxib has demonstrated non-specific targeting by inhibiting bacterial DNA and RNA replication, protein synthesis, and cell wall formation (Thangamani et al., 2015b), as well as reducing the levels of IL-6, TNF-α, IL-1β, and MCP-1 (monocyte-chemoattractant protein-1) in skin lesions caused by S. aureus infection (Thangamani et al., 2015b). Moreover, this drug has exhibited synergy with several topical and systemic antimicrobials used against S. aureus, with the exception of linezolid (Thangamani et al., 2015b).
Other NSAIDs, such diflunisal in combination with diclofenac, ibuprofen and verapamil have shown antibacterial activity against S. aureus and M. tuberculosis (Dutta et al., 2007; Vilaplana et al., 2013; Gupta et al., 2015; Hendrix et al., 2016). It was reported that diflunisal did not affect the bacterial growth of S. aureus in vitro, but did inhibit their toxicity in murine and human osteoblasts in vivo (Hendrix et al., 2016). Confirmed data have been observed in mice treated with diflunisal, which have presented less cortical bone marrow destruction, although a reduction in the bacterial load was not observed (Hendrix et al., 2016). Even though bacterial growth was not compromised, diflunisal inhibited accessory gene regulator A (AgrA), a regulator protein which inhibits alpha-type phenol soluble modulins (PSMs) and may contribute to a reduction in S. aureus virulence (Hendrix et al., 2016). In the case of verapamil, it has potentiated the activity of bedaquiline, a novel drug used to treat MDR tuberculosis, against M. tuberculosis (Dutta et al., 2007; Gupta et al., 2015). Moreover, treatment with ibuprofen significantly decreased the bacterial load and increased mice survival in an experimental model of active tuberculosis (Vilaplana et al., 2013).
For Gram-negative bacteria, celecoxib and betamethasone in combination with antibiotics have demonstrated activity against different bacterial species (Artini et al., 2014; Thangamani et al., 2015b). Celecoxib has presented synergy with colistin against A. baumannii, P. aeruginosa, Escherichia coli, K. pneumoniae and Salmonella enterica serovar Typhimurium (Thangamani et al., 2015b), and betamethasone has demonstrated synergy with ceftazidime, erythromycin, and ofloxacin against P. aeruginosa and some strains of E. coli (Artini et al., 2014). Diclofenac, in turn, was found to exhibit efficacy both in vitro and in vivo against S. enterica serovar Typhimurium (Dutta et al., 2007). In the case of glatiramer acetate, a drug used in the treatment of multiple sclerosis, activity against A. baumannii, P. aeruginosa, and E. coli reference strains, and against A. baumannii and P. aeruginosa clinical isolates from bacteremia and chronic respiratory infections in cystic fibrosis patients has been observed by disruption of the biofilm formation (Christiansen et al., 2017).
Like anticancer drugs, some anti-inflammatory and immunosuppressive drugs such as aspirin, ibuprofen, and tacrolimus have shown antifungal activity against C. neoformans, C. gattii, and E. rostratum, respectively (Sun et al., 2013; Ogundeji et al., 2016). The treatment of cryptococcal cells with aspirin and ibuprofen has led to the induction of stress via activation of the high-osmolarity glycerol (HOG) pathway in C. neoformans and C. gattii, and to their death through the activation of reactive oxygen species (ROS)-mediated membrane damage (Ogundeji et al., 2016). The MICs of these drugs did not negatively affect growth or impair macrophage function; rather, they enhanced the ability of these immune cells to phagocytose cryptococcal cells (Ogundeji et al., 2016). Moreover, treatment with tacrolimus at twice its IC50 value significantly reduced the hyphae-derived CFU of E. rostratum (Sun et al., 2013).
Antipsychotic and Antidepressant Drugs Repurposed Against Bacteria and Fungi
Trifluoperazine, an antipsychotic drug, has showed therapeutic efficacy in a murine model of C. difficile infection, presenting higher survival rates than those treated with vancomycin; a decrease in inflammation and edema was also observed compared with the infected group (Andersson et al., 2016). Furthermore, together with amoxapine, trifluoperazine in combination with vancomycin protected 80% and 100% of mice, respectively, from severe oral infection caused by C. difficile (Andersson et al., 2016). Rani Basu et al. (2005) reported that the combination of two different non-antimicrobial drugs, prochlorperazine and methdilazine, may present antibacterial activity against S. aureus.
For Gram-negative bacteria, pimozide, used for the treatment of severe Tourette’s syndrome and schizophrenia, has reduced in vitro the internalization of S. enterica serovar Typhimurium and E. coli by phagocytic cells (Lieberman and Higgins, 2009). Moreover, pimozide reduced the bacterial uptake and vacuolar escape of Listeria monocytogenes in bone marrow-derived macrophages, as well as the invasion and cell-to-cell spread of the bacteria during the infection of non-phagocytic cells (Lieberman and Higgins, 2009). In addition, the drugs trifluoperazine and amoxapine were shown to be active against Yersinia pestis after screening of a library of 780 FDA-approved drugs to identify molecules which reduce Y. pestis cytotoxicity in murine macrophages (Andersson et al., 2016). These two compounds exhibited therapeutic efficacy in a murine model of pneumonic plague by Y. pestis; although the treatment was less effective when administration of the drug was delayed (Andersson et al., 2017). However, their efficacy was improved when both compounds were administered in combination with levofloxacin (Andersson et al., 2017). In addition to this study, amoxapine was reported to present therapeutic efficacy in an experimental murine model of respiratory infection caused by K. pneumoniae (Andersson et al., 2017). Finally, azathioprine, an antidepressant drug used for the treatment of Crohn’s disease, has exhibited anti-biofilm activity against P. aeruginosa and E. coli through the inhibition of WspR (Antoniani et al., 2013). WspR is a diguanylate cyclase involved in the regulation of a signal molecule called cyclic-di-GMP (c-di-GMP) known as a regulator of the bacterial biofilm formation (Antoniani et al., 2013).
In the case of fungi, the antipsychotic drug bromperidol has exhibited synergy with various azoles against C. albicans, C. glabrata, and Aspergillus terreus (Holbrook et al., 2017). Bromperidol has demonstrated synergy with posaconazole and voriconazole, and partial synergy with itraconazole and ketoconazole against C. albicans, C. glabrata, and A. terreus, as demonstrated by checkerboard and time-kill assays (Holbrook et al., 2017). Moreover, bromperidol in combination with posaconazole and voriconazole, increased the disruption of biofilm formation by sessile cells of C. albicans induced by both azoles. Their sessile MICs were reduced from >32 to 0.5 mg/L (Holbrook et al., 2017).
Other Drugs Repurposed Against Bacteria and Fungi
Other drugs with different modes of action and clinical indications have been evaluated as antibacterial agents. Auranofin, which is used for the treatment of rheumatoid arthritis, has shown in monotherapy greater activity against a wide range of Gram-positive bacteria including S. pneumoniae, S. aureus, Enterococcus faecium, E. faecalis, and Streptococcus agalactiae when compared with vancomycin or linezolid (Aguinagalde et al., 2015; Thangamani et al., 2016a,b). In vivo, auranofin and its analogs have demonstrated therapeutic efficacy in different experimental models such as MRSA septicemic infection, MRSA skin infection, MRSA implant infection model (a model involving mesh-associated biofilm), and MRSA intramuscular infection model (Aguinagalde et al., 2015; Thangamani et al., 2016a,b). Interestingly, auranofin has demonstrated synergy with the commonly used antibiotics such as ciprofloxacin, linezolid, and gentamicin against MRSA (Thangamani et al., 2016b). In order to improve the activity of auranofin, different analogs were synthetized and display improved antibacterial activity against S. aureus and S. pneumoniae causing bacteremia in murine model (Aguinagalde et al., 2015). The mode of action of auranofin against S. aureus has been deciphered using the macromolecular biosynthesis assay which showed that auranofin acts on the inhibition of DNA replication and protein synthesis, downregulating the toxin production (Thangamani et al., 2016a).
Ebselen; despite the fact that it is not an FDA-approved drug, it is being investigated in clinical trials for the treatment of bipolar disorder and ischemic stroke, has also been evaluated. Two studies have suggested that this compound exhibited antibacterial activity against C. difficile in vitro and in vivo by targeting glucosyltransferase domain (GTD) of C. difficile toxins (Peng et al., 2018), and against MRSA and vancomycin-resistant S. aureus (VRSA) with MIC values <1 mg/L (Thangamani et al., 2015c). Moreover, ebselen has also reduced the biofilm formation by S. aureus (Gi et al., 2014). Synergy between this drug and fusidic acid, retapamulin, mupirocin, and daptomycin against S. aureus strains was confirmed using a Bliss model (Thangamani et al., 2015c).
Besides these two drugs, the antihistaminic compounds terfenadine and its analogs were also investigated as potential antibacterial drugs. Terfenadine has showed reasonable activity against S. aureus (Perlmutter et al., 2014). In order to improve their activity, 84 derivatives were synthesized that have presented greater MIC values against S. aureus (1 mg/L) as well as activity against E. faecium, E. faecalis, and M. tuberculosis (Perlmutter et al., 2014).
In the case of statins, simvastatin, used in the treatment of atherosclerotic cardiovascular disease and hypercholesterolemia, has exhibited antibacterial activity in monotherapy against M. tuberculosis (Parihar et al., 2014; Skerry et al., 2014). It marginally reduced the bacterial load 4 and 8 weeks after infection with M. tuberculosis by aerosol exposure; presumably by reducing cholesterol synthesis due to the inhibition of HMG-CoA reductase within the phagosomal membrane. This process has consequently enhanced the maturation of phagosomes, known to provide better defense against M. tuberculosis, and by inducing the autophagy of M. tuberculosis-infected macrophages (Parihar et al., 2014). The addition of simvastatin to the first-line tuberculosis therapy (rifampicin + isoniazid + pyrazinamide) may help to reduce mycobacterial infection and tissue damage in M. tuberculosis-infected mice (Skerry et al., 2014). Similarly, atorvastatin, another statin drug, has also demonstrated synergistic activity with rifampin in vitro against M. tuberculosis and in a murine model of Mycobacterium leprae infection (Lobato et al., 2014).
Regarding Gram-negative bacteria, auranofin exhibited synergy with polymyxin B against A. baumannii, P. aeruginosa, K. pneumoniae and S. enterica serovar Typhimurium (Thangamani et al., 2016a).
Ebselen has also presented antibacterial effect against A. baumannii and E. coli by reducing their bacterial growth at MICs of 32 μM and <128 μM, respectively. This bacterial reduction growth was due to the inhibition of TonB-mediated physiology, which is involved in iron acquisition from host sources (Nairn et al., 2017). Furthermore, ebselen exhibited anti-virulence activity against P. aeruginosa by targeting c-di-GMP signaling pathway, which regulates motility and biofilm formation (Gi et al., 2014; Lieberman et al., 2014).
In the case of statins, the combination of simvastatin with sub-inhibitory concentrations of colistin presented synergistic effect against a collection of A. baumannii, E. coli, K. pneumoniae, P. aeruginosa, and S. enterica serovar Typhimurium reducing the MIC of simvastatin from >256 mg/L to a range between 8 and 32 mg/L (Thangamani et al., 2015a). In addition, screening of an FDA-approved drug library has identified pentetic acid, an iron chelator, as an inhibitor of elastase, an important exoprotease as well as a reducer of biofilm formation by P. aeruginosa (Gi et al., 2014). When applied to P. aeruginosa infections in human lung tissue, pentetic acid increased the viability of human lung epithelial A549 cells post-infection (Gi et al., 2014). Interestingly, pentetic acid has also demonstrated therapeutic efficacy in a murine experimental model of respiratory infection by P. aeruginosa by increasing 42% the mice survival 5 days post-infection (Gi et al., 2014).
Moreover, calcitriol, a bioactive form of vitamin D3 used to treat hypocalcemic conditions and renal osteodystrophy, has been described as an enhancer of bactericidal activity against P. aeruginosa, due to its capacity to modulate the activity of monocytes and macrophages by increasing their bacterial killing (Nouari et al., 2015).
Other drugs that have presented anti-virulence effect against P. aeruginosa have been reported. For example 5-fluorocytosine, an antifungal drug, has been shown to reduce in vitro the production of virulence factors by P. aeruginosa such as pyoverdine, PrpL protease, and exotoxin A by downregulating pvdS gene expression (Imperi et al., 2013a), and to suppress in vivo the pathogenicity of P. aeruginosa in a murine model of lung infection (Imperi et al., 2013a). Other antifungal drugs such as clotrimazole and miconazole were identified as inhibitors of 2-heptyl-3-hydroxy-4 quinolone (PQS) quorum sensing (QS) system. This system is based on signal 2-alkyl-4-quinolones (AQ): PQS and 2-heptyl-4-hydroxyquinolone (HHQ) which can bind and activate the regulator PqsR and controls the expression of P. aeruginosa virulence factors. D’Angelo et al. (2018) have shown that probably both drugs modify PqsR function by competing with PQS and HHQ for the PqsR ligand-binding site. Finally, clofoctol and azithromycin, drugs originally developed as antibiotics against Gram-positive and Gram-negative bacteria, respectively, were found to have also anti-virulence properties against P. aeruginosa (Imperi et al., 2014; D’Angelo et al., 2018).
In the case of fungi, atorvastatin has demonstrated different effects on the host and the yeast by: (i) reducing the ergosterol content in the cell membrane and altering the properties of the polysaccharide capsule of C. gattii; (ii) increasing the production of ROS by macrophages; and (iii) reducing yeast phagocytosis and the intracellular proliferation rate (Ribeiro et al., 2017). Atorvastatin in combination with fluconazole was also tested as an adjuvant to control fungal infections. This combination demonstrated synergy in vitro against one strain of C. gattii. In vivo, atorvastatin plus fluconazole increased the survival of mice and reduced the burden of C. gattii in the lungs and brain (Ribeiro et al., 2017). Moreover, preclinical antimalarial drugs such as MMV665943 have been shown to inhibit and delay growth at submicromolar concentrations and exhibit fungicidal activity at concentrations greater than 1.56 μM against C. albicans, C. neoformans, C. gattii and Lomentospora prolificans. More specifically, this compound at concentrations greater than 1.56 μM affects the polysaccharide capsule thickness of C. neoformans (Jung et al., 2018).
Regarding the immune response modulation, ebselen and auranofin reduced the production of inflammatory cytokines such as TNF-α, IL-6, IL-1β, and MCP-1 in skin lesions infected by S. aureus (Thangamani et al., 2016a, 2015c). Similarly, calcitriol has shown a modulatory effect on monocytes and macrophages against P. aeruginosa infection by increasing their bacterial killing (Nouari et al., 2015). The mechanism of action of calcitriol on the immune system is unknown; although its downregulating effect on IL-1β, IL-6, and IL-8 has been observed (Xue et al., 2002). In the case of statin, simvastatin has been reported to modulate the production of proinflammatory cytokines (IL-8 and CCL20) and Kruppel-like factors (an emerging group of immune system regulators) in P. aeruginosa respiratory infections (Hennessy et al., 2014).
Clinical Application of Repurposed Drugs Against Infectious Agents
Even though repurposed drugs showed promising preclinical data, to our knowledge only three clinical studies have been performed or are currently underway.
A randomized study on the role of aspirin in tuberculous meningitis suggested that aspirin in combination with corticosteroids reduced the incidence of strokes and mortality (Misra et al., 2010). A similar study on the role of aspirin as an adjunct with steroids for the treatment of HIV-negative adults with tuberculous meningitis in Vietnam is still ongoing, now in Phase II trial (clinical trials identifier: NCT02237365). Another Phase III trial (ClinicalTrials.gov Identifier: NCT02060006) is being conducted to evaluate the feasibility and efficacy of using meloxicam, a cheap and widely available NSAID, as a preventive intervention for tuberculous-immune reconstituted inflammatory syndrome; results from this study have yet to be published (Maitra et al., 2016).
Conclusion and Perspectives
In the last decade, substantial progress has been made in the development of repurposed drugs for the treatment of bacterial and fungal infections. Several compounds have yielded promising data but developmental efforts remain in the preclinical stage. Additional relevant issues should be take into account in the preclinical development of repurposing drugs including possible need for new formulations to increase their bioavailability and ADMET tests if the administration route is changed, possible negative effect of the primary drug activity (especially for anticancer and antipsychotic drugs), and challenges for intellectual property rights. Moreover, further clinical studies are needed to address the urgent demand for new treatments targeting infections caused by bacteria and fungi.
More information: Gholamreza Haqshenas et al. Targeting of host cell receptor tyrosine kinases by intracellular pathogens, Science Signaling {2019) DOI: 10.1126/scisignal.aau9894
Journal information: Science Signaling



{kind=link}