")










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Multiple sclerosis is an autoimmune disease affecting both adults and children.
It’s driven by “helper” T cells, white blood cells that mount an inflammatory attack on the brain and spinal cord, degrading the protective myelin sheath that covers nerve fibers. But there are many different kinds of T helper cells, and up until now, no one knew which ones were the bad actors.
Researchers at Boston Children’s Hospital have now pinpointed the specific helper T cells that cause MS, as well as a protein on their surface that marks them.
As reported this week in PNAS, an antibody targeting this protein, CXCR6, both prevented and reversed MS in a mouse model.
If human studies bear out the findings, targeting these rogue T cells could potentially ameliorate MS, says senior investigator Eileen Remold-O’Donnell, PhD, of the hospital’s Program in Cellular and Molecular Medicine.
She believes the findings could also apply to other forms of autoimmune encephalomyelitis (inflammation of the brain and spinal cord), as well as inflammatory arthritis.
“We’ve demonstrated in mice you can target these cells and get rid of them,” she says. “We don’t know if this approach would be appropriate for all cases of MS, but it could be effective in the early inflammatory stages of the disease, and in targeting newly formed cells during disease exacerbations.”
Profiling MS-inducing cells
Recent efforts to pinpoint the T helper cells causing MS have pointed to TH17 cells, but some TH17 cells appear not to be involved. Remold-O’Donnell and her former postdoctoral fellow Lifei Hou, PhD, zeroed in on a subset of fast-proliferating TH17-derived cells, all bearing the CXCR6 marker.
These cells, they showed, are highly damaging to nerve fibers, producing one set of proteins that directly damage cells and others, including GM-CSF, that stimulate an inflammatory attack by other immune cells known as macrophages.

The CXCR6-positive cells also produce increased amounts of a protein called SerpinB1 (Sb1), the researchers showed. When they deleted the Sb1 gene in T cells in their mouse model, fewer immune cells survived to infiltrate the spinal cord. The mice also displayed fewer symptoms of disease than control mice.
Moreover, these Sb1-containing cells could be readily identified with antibodies targeting the CXCR6 surface protein.
Human counterparts
But are CXCR6+ cells relevant in human disease?
To investigate, Remold-O’Donnell and Hou worked with rheumatologists, immunologists, and neuro-immunologists at Boston Children’s and Brigham and Women’s Hospital.
They obtained and tested samples of blood and synovial fluid (from the cavities of joints) from patients with inflammatory autoimmune arthritis. Levels of CXCR6+ cells were indeed elevated in the inflamed joints, but not in circulating blood from the arthritis patients, patients with MS, or healthy controls.
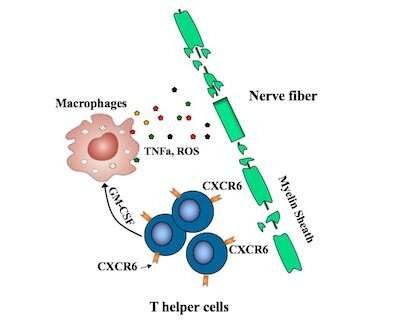
Targeting CXCR6 in multiple sclerosis
Remold-O’Donnell and Hou, first author on the paper, believe treatments to deplete CXCR6+ cells could mitigate MS and possibly other autoimmune disorders while largely leaving other T cell immune defenses intact.
When they used monoclonal antibodies to target CXCR6, the harmful cells largely disappeared, and mice, which were primed to get MS, did not develop the disease.
The two have filed a patent covering the work and have formed a company, Edelweiss Immune, Inc., in which they have equity ownership together with Boston Children’s Hospital. The new company will be carrying the research forward.
“Many drugs have been developed to treat autoimmune diseases, such as glucocorticoids and cytotoxic reagents,” says Hou. “However, none selectively target pathogenic T cells, and long-term use of immunosuppressive agents results in broad immunosuppression and compromised immune defenses. Therapeutics with better selectivity, safety, and efficacy are needed.”
Multiple sclerosis
Overview
Multiple sclerosis (MS) is an inflammatory disorder of the brain and spinal cord characterized by focal lymphocytic infiltration and microglial activation leading to neurodegeneration and progressive disability [1].
MS is the most common chronic neurological disease in young and middle-aged adults, affecting 2.5 million people worldwide. It is more prevalent in Northern Europe, Canada and Oceania and shows a female preponderance, with a female-to-male 2:1 ratio. MS is classified into three subtypes, namely relapsing–remitting MS (RRMS), primary progressive MS (PPMS) and secondary progressive MS (SPMS). RRMS, which accounts for 80% of the patients, presents at first with an acute episode affecting one or more sites, known as the clinically isolated syndrome (CIS). A second attack of demyelination occurring afterward is required to meet the diagnostic criteria for RRMS. Ultimately, around 65% of RRMS patients enter the SPMS phase, while in 20% the illness is progressive from onset, hence the characterization as PPMS [1].
The first therapeutic regimens that became available for MS were interferon-β and glatiramer acetate [2–4]. The FDA has currently licensed several immune disease-modifying therapies (DMTs), most of which have been validated in other autoimmune diseases as well. These include monoclonal antibodies, such as rituximab (anti-CD20) [5] and alemtuzumab (anti-CD52) [6, 7], and oral agents with immunomodulatory properties, such as fingolimod, dimethyl fumarate and teriflunomide [8–10]. Clinical trials testing anti-CD20-mediated depletion of peripheral B cells showed promising effects against the development of new central nervous system (CNS) lesions and relapses [11, 12]. Despite some efficacy of therapies, there are still unmet therapeutic needs in MS. Besides, current therapeutic agents are costly summing up to a total annual cost of approximately 15 billion euros for MS in Europe in 2010.
Pathogenesis
Environmental, genetic, epigenetic and immunological factors are implicated in the development of MS [13, 14]. Myelin-targeting autoreactive CD4(+) T cells that pass through a disrupted blood–brain barrier (BBB) and enter the CNS were initially considered the critical orchestrators in the pathogenesis of MS [15]. Activated by microglia, astrocytes or other immune cells through HLA class II presentation of myelin antigens, CD4(+) T cells express different cytokines depending on their subset. Th1 and Th17 cells express pro-inflammatory cytokines, such as IFN-γ and IL-17, respectively, whereas Th2 and regulatory T cells (Tregs) produce anti-inflammatory cytokines, such as IL-10 [16–18]. Thus, the skewing toward Th1 and Th17 responses is responsible for the immune-mediated damage of myelin and axons [19].
The activation of CD4(+) T cells within the CNS leads to the recruitment of other inflammatory cells, such as B cells, which cross the BBB, undergo activation, antigen-driven affinity maturation and clonal expansion [20]. In recent years, accumulated evidence emphasizes the role of B cells in the progression of MS [21, 22]. B cells are especially efficient in presenting antigens to CD4(+) T cells through HLA class II molecules [23]. Apart from antigen presentation, B cells are also able to produce autoantibodies after differentiation to plasma cells. Autoantibodies are able to cause demyelination through antibody-dependent cellular cytotoxicity (ADCC) and complement activation. Lastly, B cells are able to express pro-inflammatory cytokines, such as IL-6, IFN-γ and TNFα, known to be implicated in MS [24–26]. In particular, RRMS patients were found to have elevated peripheral expression of IFN-γ, interleukin (IL) 1-beta (IL1B), IL-7, IL-10, IL12A, IL-15, IL-23, IL-27, lymphotoxin-alpha (LTA) and lymphotoxin-beta (LTB) [27]. Τhe main sources of pro-inflammatory cytokines within PBMCs were T- and B cells, whereas monocytes were the most noticeable source of immunoregulatory cytokines [27]. Hence, the inflammatory reaction of T, B and other immune cells leads to demyelinated lesions throughout the CNS [28]. Interestingly, healthy individuals also possess autoreactive T cells at lower frequencies, a finding which signifies that their presence is not enough per se for disease induction [29]. Thus, healthy individuals are likely to maintain regulatory mechanisms that keep these autoreactive T cells under control. Emerging evidence suggests that both Tregs and Bregs play a major role in this ‘safeguard’ process.
Tregs
Tregs were originally identified by Sakaguchi et al. in 1995 as a CD4(+)CD25(+) T cell subset with suppressive activity [30]. They are essential to the maintenance of self-tolerance and their impairment has been linked with autoimmunity and includes numerical decreases, functional defects and conversion into inflammatory effector cells [31]. High expression of CD25 and low expression of CD127 are the main phenotypic markers characterizing bona fide human Tregs [32]. CD25 (IL-2 receptor) is central to Treg ontogeny, optimal regulatory function and proliferation mediated by the gamma chain cytokines IL-2, IL-4, IL-7 and IL-15 [33]. Subsequently, it was shown that cells with regulatory capacity can also express CD8 [34], cytotoxic T-Lymphocyte antigen-4 (CTLA-4) [35, 36], the TCR-inducible co-stimulatory receptor (ICOS) [37] and high levels of CD5 [38, 39], a surface marker that instructs extrathymic Treg cell development in response to self and tolerizing antigens also co-expressed by certain B regulatory cell subsets [40, 41] (as discussed below). The seminal discovery of forkhead box P3 (FoxP3), as a fundamental transcription factor for the development of regulatory CD4(+)CD25(+) T cells in the thymus, helped researchers to precisely phenotype most Tregs [42]. FoxP3 (also known as Scurfin, IPEX, and JM2) is a transcriptional repression factor of the winged helix family and is expressed in all CD4(+) Treg cells with regulatory activity. Currently, Tregs may be accurately identified as CD4(+)CD25(+)FoxP3(+) T cells or (as FoxP3 inversely correlate with cell surface CD127 expression) as CD4(+)CD25(+)CD127(lo)/(−) T cells [43]. Specific regulatory T cell populations may also express other surface markers such as CD39, LAG-3 and GITR [44–47].
Natural and induced Tregs
Tregs can be subdivided into thymus-developed, “natural” Tregs that mediate tolerance to self-antigens and “induced” Tregs derived from conventional CD4(+) T cells following non-self antigenic exposure [48]. Natural Treg production requires stable expression of FoxP3 and high-affinity binding of HLA/self-peptide complex on thymic antigen-presenting cells (APCs) to T cell receptor (TCR). Natural Tregs can be also sub-classified into CD45RA(+)“naïve” Tregs and CD45RO(+)“memory” Tregs [49]. ‘Induced’ or ‘adaptive’ Tregs (iTregs) are generated from naïve T cells in the presence of transforming growth factor-β (TGF-β) or retinoic acid and produce the anti-inflammatory cytokine IL-10 [50–52]. Despite the phenotypic and functional overlaps with natural Tregs, iTregs demonstrate apparent differences in stability and gene expression [53]. Type 1 regulatory T cells (Tr1), are a subpopulation of Tregs-expressing CD4(+)CD49(+)LAG-3(+)IL-10(+) that exert significant immunosuppressive effects [54–56]. In addition, CD8(+) Tregs (Tr2) and IL-17-producing Tregs that share some common features with Tr1 also exist [57]. iTregs-expressing RORγt, which is the master regulator of antimicrobial type 3 immunity are termed type 3 Tregs (Tr3) [58, 59]. These cells constitute the major population of colonic Tregs, require bacterial antigens for differentiation and are distinct from thymus-derived Tregs.
Tregs function
Tregs have a pivotal function in regulating the immune system by controlling the number and function of effector cells. Thus, they play a major role in suppressing unwanted autoreactive immune responses, such as in the case of autoimmunity [60]. Interestingly, it has been indicated that Tregs can modulate both adaptive and innate immune systems, and once activated they specifically regulate immune responses at multiple levels and by various mechanisms. These suppressive mechanisms can be organized into major groups, including cell–cell contact-dependent suppression, inhibitory cytokine release (such as IL-10 and TGF-β), modulation of APC function, cytolysis, metabolic disruption and induction of suppressor cells or “infectious tolerance” [53].
In addition to IL-10, the inhibitory cytokine IL-35 also contributes to regulatory T cell function [61, 62]. IL-35 belongs to IL-12 family of cytokines that includes IL-12, IL-23, IL-27 and IL-35. Of these, IL-12 and IL-23 have pro-inflammatory roles, whereas IL-35 appears to exert a more regulatory function by inducing the expansion of Tregs and Bregs subsets and inhibiting Th17 cell differentiation [63]. IL-35-producing Tregs represent a distinct effector population from the IL-10-producing iTregs which also have different transcription factor dependency, as differentiation regulator Blimp1 is essential for IL-10 production, but not for IL-35, whereas Foxp3 is important for IL-35 but dispensable for IL-10 production [64]. Recently, it was demonstrated that the IL-12p35 alpha subunit of IL-35 efficiently suppressed encephalitogenic T cell responses and ameliorated experimental autoimmune encephalomyelitis (EAE), a well-characterized murine model of MS [65]. IL-12p35 inhibited the expansion of pathogenic Th17 and Th1 cells and mediated the expansion of Tregs and Bregs [65].
Tregs and multiple sclerosis
Major studies investigating the role of Tregs in MS are summarized in Table 1.
Table 1
Main studies investigating the effect of MS-treated patients on regulatory B and T cells
| Authors, year of study | Origin/country | Treatment | Sample | Results |
|---|---|---|---|---|
| Quan et al. (2015) | China | Rituximab | Healthy controls (n = 19) NMO patients (n = 9) | Tregs increased from 0.3 to 1.2% of total lymphocytes after 48 weeks |
| De Mercanti et al. (2016) | Europe | Alemtuzumab | RRMS patients (n = 29) | Significant increase in CD4(+)CD25(hi)CD127(lo)FoxP3(+) Tregs after 24 months of treatment |
| Haas et al. (2015) | Germany | Fingolimod | Healthy controls (n = 37) MS patients (n = 74) | Increased median percentage of Tregs from 3 to 6,7% after 3 months of treatment |
| Blumenfeld et al. (2016) | Israel | Fingolimod | MS patients (n = 10) | Increase in the percentage of CD38(hi)CD24(hi) “transitional” Bregs from 3.7 to 11.6% |
| Piancone et al. (2016) | Italy | Fingolimod | RRMS patients (n = 12) | Significant increase in CD19(+)BTLA(+)IL-10(+) B cells both as a percentage of total lymphocytes and CD19(+) B cells |
| Lundy et al. (2016) | USA | Dimethyl Fumarate | RRMS patients (n = 13) | After 12 months of treatment: CD19(+) B cells concentration was halved and CD24(hi)CD38(hi) Bregs were doubled |
| Stenner et al. (2008) | Germany | Natalizumab | RRMS patients (n = 15) | No significant change in Tregs percentage 30 days after initiation of therapy |
| Putzki et al. (2010) | Switzerland | Natalizumab | RRMS patients (n = 28) | Relative decrease in CD4(+)CD25(+) Tregs from 18.9 to 14.1% |
| Schubert et al. (2015) | USA | IFN-β | Treatment-naïve RRMS patients (n = 10) IFN-β-treated RRMS patients (n = 11) | Increase in CD24(hi)CD38(hi) “transitional” Bregs from 1.09 to 9.50% |
| Ireland et al. (2014) | USA | Glatiramer acetate | Treatment-naïve MS patients (n = 22) Glatiramer acetate-treated MS patients (n = 22) | Treated patients IL-10 production by B cells was equivalent to those in healthy donors and up to 6.5-fold greater than the levels in treatment-naive patients |
Conclusion
Multiple sclerosis is the most prevalent autoimmune disease of the CNS and a frequent cause of neurological disability in young adults.
As there is no cure for the disorder, the aim of new treatments is the alleviation of symptoms and the reduction of relapses.
As with most autoimmune diseases, MS patients exhibit impaired immunoregulatory mechanisms that lead to harmful immune responses. It is not yet recognized whether this dysregulation is the cause or a consequence of the disease. Nevertheless, regulatory mechanisms play a major role in MS.
Thus, it comes as no surprise that most if not all of MS therapies have immunomodulatory actions.
It is important to conduct more research on current medications and their influence on regulatory lymphocytes to uncover their exact mechanism of action and to be able to administer the appropriate therapeutic agent to each patient, according to their particular condition (personalized or precision medicine).
On a final note, other agents that are not currently in use in MS but have immunomodulatory properties, such as vitamin D or statins, could be beneficial as a complementary treatment for MS.
More information: Lifei Hou et al. SerpinB1 controls encephalitogenic T helper cells in neuroinflammation, Proceedings of the National Academy of Sciences (2019). DOI: 10.1073/pnas.1905762116
Journal information:Proceedings of the National Academy of Sciences



{kind=link}