











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Tau can quickly spread between neurons but is not immediately harmful, according to research in mouse neurons published in Journal of Neuroscience.
Intervening during the initial accumulation of tau could potentially halt the progression of Alzheimer’s disease.
A hallmark of Alzheimer’s disease is the accumulation of tau protein in neurons, which leads to their death.
A diseased version of tau folds itself incorrectly, which leads to the buildu
Researchers did not know the timescale of this process and how misfolded tau can spread to other cells.
A functioning brain cell that is expressing diseased tau.
Hallinan et al. introduced diseased tau into mouse neurons growing in a cell culture. Within days, the activated tau had spread to other neurons and began misfolding and accumulating.
Despite the tau buildup, both the donating and accepting neurons remained healthy and capable of sending electrical messages.
These results show that tau buildup itself is not harmful, but rather it is the cellular processes it disrupts that kill neurons.
Alzheimer’s disease (AD) is an irreversible progressive neurological disorder that is characterized by memory loss, the retardation of thinking and reasoning, and changes in personality and behaviors.1,2 AD seriously endangers the physical and mental health of the elderly. Aging is the biggest risk factor for the disease, the incidence of which doubles every 5 years after the age of 65.3 Approximately 40 million people over the age of 60 worldwide suffer from AD, and the number of patients is increasing, doubling every 20 years.4,5,6,7
In 1906, Alois Alzheimer presented his first signature case and the pathological features of the disease at the 37th convention of Southwestern German Psychiatrists.
Later, in 1910, his coworker Emil Kraepelin named the disease in honor of his achievements. In the following years (from 1910 to 1963), researchers and physicians did not pay much attention to the disease until Robert Terry and Michael Kidd revived interest by performing electron microscopy of neuropathological lesions in 1963.
Electron microscopy analysis showed that neurofibrillary tangles (NFTs) were present in brain biopsies from two patients with advanced AD.8,9 Since then, studies on the pathological features and mechanisms of AD and drug treatments for the disease have been conducted for more than half a century (from 1963 to present).10
Clinically, AD is divided into sporadic AD (SAD) and familial AD (FD). FD accounts for 1–5% of all AD cases.11,12,13,14,15
In the early 1990s, linkage analyses of early-onset FD determined that mutations in three genes, namely, amyloid-beta A4 precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), are involved in FD. PSEN1 mutations account for ~81% of FD cases, APP accounts for ~14%, and PSEN2 accounts for ~6%.11 In addition to these three genes (APP, PSEN1, and PSEN2), more than 20 genetic risk loci for AD have been identified.16,17
The strongest genetic risk factor for AD is the ε4 allele of apolipoprotein E (APOE).18,19,20,21 APOE is a class of proteins involved in lipid metabolism and is immunochemically colocalized to senile plaques, vascular amyloid deposits, and NFTs in AD. The APOE gene is located on chromosome 19q13.2 and is associated with late-onset FD. The APOE gene has three alleles, namely, ε2, ε3, and ε4, with frequencies of 8.4%, 77.9%, and 13.7%, respectively.
The differences in APOE2 (Cys112, Cys158), APOE3 (Cys112, Arg158), and APOE4 (Arg112, Arg158) are limited to amino acid residues 112 and 158.22,23,24,25 Analyses of the frequencies of these APOE alleles among human populations have revealed that there is a significant association between APOE4 and late-onset FD (with an ε4 allele frequency of ~40% in AD), suggesting that ApoE4 may be an important susceptibility factor for the etiopathology of AD.25,26,27
Moreover, APOE4 can increase the neurotoxicity of β-amyloid (Aβ) and promote filament formation.28 The APOE4 genotype influences the timing and amount of amyloid deposition in the human brain.29 Reelin signaling protects synapses against toxic Aβ through APOE receptors, which suggests that APOE is a potential target for AD therapy.30
The incidence of SAD accounts for more than 95% of all AD cases. Therefore, in this review, we focus our attention on recent SAD research and clinical trials.
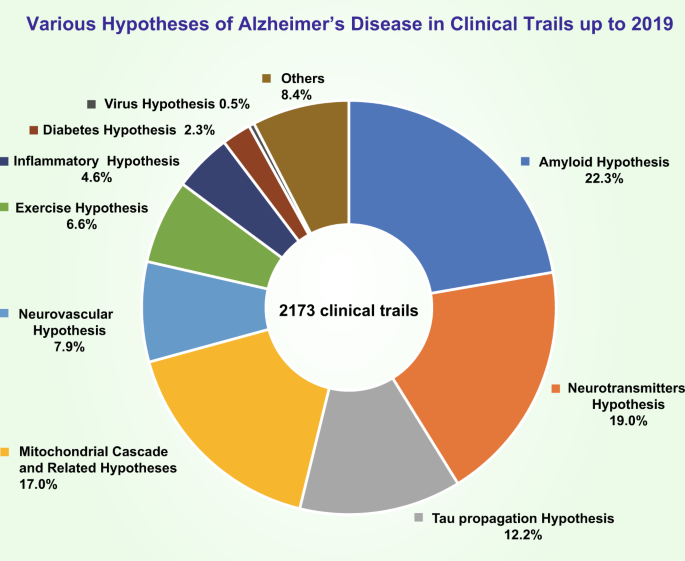
There are various descriptive hypotheses regarding the causes of SAD, including the cholinergic hypothesis,31 amyloid hypothesis,32,33 tau propagation hypothesis,34 mitochondrial cascade hypothesis,35 calcium homeostasis hypothesis,36 inflammatory hypothesis,37 neurovascular hypothesis,38 metal ion hypothesis,39 and lymphatic system hypothesis.40 In addition, there are many other factors that increase the risk for SAD, including family history,41 midlife hypertension,42 sleep disorders,43 midlife obesity,44 and oxidative stress.45,46 Interestingly, according to the latest evaluation of single-nucleotide polymorphisms (SNPs), Mukherjee et al. found 33 SNPs associated with AD and assigned people to six cognitively defined subgroups.47
At present, clinical drug treatments are mainly divided into two categories: acetylcholinesterase inhibitors (AChEIs), represented by donepezil, and the antagonist of N-methyl-D-aspartic acid (NMDA) receptor, represented by memantine (Table 1).48
As neurotransmitter regulators, these drugs can only relieve symptoms for a short time but cannot delay the progression of AD. Recent failures and the limited progress of therapeutics in phase III clinical trials suggest that it is time to consider alternative strategies for AD treatment.49
In this review, we discuss the hypotheses of the molecular mechanisms of AD and related clinical trials (Fig. 1) and hope that these discussions will be helpful for developing explanatory theories and potential effective strategies for AD treatment.
Source:
SfN
Media Contacts:
Calli McMurray – SfN
Image Source:
The image is credited to Hallinan et al., JNeurosci 2019.
Original Research: Closed access
“Tau Misfolding Efficiently Propagates Between Individual Intact Hippocampal Neurons”. Grace I Hallinan, Mariana Vargas-Caballero, Jonathan West and Katrin Deinhardt.
Journal of Neuroscience doi:10.1523/JNEUROSCI.1590-19.2019.



{kind=link}