










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Aggregates of the protein alpha-synuclein arising in the gut may play a key role in the development of Parkinson’s disease (PD).
Investigators are testing the hypothesis that by targeting the enteric nervous system with a compound that can inhibit the intracellular aggregation of alpha-synuclein, they can restore enteric functioning in the short term, and possibly slow the progressive deterioration of the central nervous system in the long term.
They review results to date in the Journal of Parkinson’s Disease.
“There is growing evidence that PD may start off in the gut,” explained senior author Michael Zasloff, MD, PhD, Enterin Inc., Philadelphia, PA, and MedStar Georgetown Transplant Institute, Georgetown University Medical Center, Washington, DC.
“The concept is that aggregates of the protein alpha-synuclein, thought to play a key role in the disease, arise within the enteric nervous system (ENS) and travel up the peripheral nerves to the central nervous system (CNS) where they ultimately cause inflammation and destruction of parts of the brain.
Targeting the formation of alpha-synuclein aggregates in the ENS may therefore slow the progression of the disease.”
Alpha-synuclein is one of the defensive proteins produced by enteric nerves when they encounter infections. In children with acute bacterial GI infections, for example, intestinal nerves produce alpha-synuclein.
In children who have undergone intestinal transplants and who are prone to GI infections, investigators have shown that enteric neurons start making alpha-synuclein at the time of acute viral infections, and this outlasts the infection by many months, protecting nerve cells for prolonged periods of time.
Within a nerve cell, alpha-synuclein could envelop invading viruses and disrupt their replication. It could also attach itself to small vesicles containing neurotransmitters and be released from the nerve cell hitching a ride with them. Once on the outside, it can attract protective immune cells from surrounding tissues.
“Recent data from our laboratory and others demonstrate that alpha-synuclein is induced in the setting of viral and bacterial infection and serves an immune function by protecting the ENS, by alerting the adaptive immune system and through pre-emptive defense of the CNS in advance of the infectious agent,” noted first author Denise Barbut, MD, FRCP, Enterin Inc., Philadelphia, PA.
“In the setting of chronic GI infections or impaired intestinal barrier function, when the expression of alpha-synuclein exceeds its clearance, neurotoxic aggregates of alpha-synuclein form damaging aggregates in the ENS and traffic to the CNS.”
To determine whether targeting alpha-synuclein within enteric neurons might help patients with PD, Dr. Barbut, Dr. Zasloff and colleagues are currently conducting clinical trials with a compound called ENT-01 (Enterin Inc.).
ENT-01 is a synthetic derivative of squalamine, a compound originally isolated from dogfish bile by Dr. Zasloff.
It displaces alpha-synuclein from nerve cell membranes and restores the normal electrical activity of enteric neurons.
Investigators completed a 50-patient Phase 2a study (RASMET) in patients with PD in 2018, which corrected constipation in more than 80% of participants, with the dose titrated up for each patient until a response was obtained. Constipation is a common symptom of PD.

This is a graphic representation of the proposed immune roles of alpha-synuclein within the ENS. The image is credited to University of Groningen – The Netherlands.
According to Dr. Barbut, “The RASMET study demonstrated that the ENS is not irreversibly damaged in patients with PD, despite the longstanding constipation that might suggest otherwise.
We believe that this is the first demonstration of the reversal of a neurodegenerative process in humans.” Beyond the bowel symptoms, possible benefits were also observed in motor and non-motor symptoms such as hallucinations, depression and cognitive function.
A 110-patient double-blind, placebo-controlled Phase 2b trial (KARMET) evaluating the effect of oral ENT-01 tablets on constipation and neurologic symptoms is currently being conducted.
PD is a slowly progressive disorder that affects movement, muscle control and balance. It is the second most common age-related neurodegenerative disorder affecting about 3% of the population by the age of 65 and up to 5% of individuals over 85 years of age.
Parkinson’s Disease (PD) is a progressive neurodegenerative disease, which accounts for approximately 15% of all dementia cases [1], and is the second most common form of neurodegeneration to Alzheimer’s disease [2]. The disease has a mean onset of 55 years old and exhibits both physical and neuropsychiatric symptoms.
The physical symptoms include slow imprecise movements (bradykinesia), tremors at rest, rigidity, facial paucity (hypomimia), shuffling gait, difficulty walking, freezing and postural instability [2]. The neuropsychiatric symptoms, which occur at later stages of the disease, manifest as cognitive defects, specifically slowness, disrupted sleep, and sensory disturbances, leading to suffers becoming passive and withdrawn [2].
PD is thought to be largely caused by the death of dopaminergic neurons in the substantia nigra pars compacta, located in the basal ganglia of the brain. This region of the brain is involved in coordinating movement, sending signals down the spinal cord to control muscle contraction, meaning that damage to this region can compromise signalling, leading to the physical symptoms of PD.
A wide range of both environmental and genetic risk factors have been implicated in the pathogenesis of PD [3]. Environmental risk factors include pesticides (specifically organochlorines) [4] and ambient air pollution [5]. Interestingly, tobacco [6], coffee [7], black tea [8], and a few pharmaceuticals including statins [9], calcium channel blockers [10] and ibuprofen [11], have shown some evidence of neuroprotective properties in a few studies. Autosomal dominant risk factors implicated with PD were first found in the SNCA gene that encodes αS, the primary component of Lewy bodies that are characteristic of all synucleinopathies.
This will be discussed in detail and is the main focus of this review. It is worth noting that there are a number of other autosomal dominant and recessive risk factors implicated in PD, some of which occur upstream of the toxicity caused by αS. Other autosomal dominant mutations are found in the Leucine rich repeat Kinase 2 (LRRK2) domain, accounting for 4% of familial PD [12], in the vascular protein sorting 35 (VPS35) gene [13], accounting for 1% of familial PD and in the CHCHD2 [14] and eIF4G1 [15] genes. Recessive genes implicated in familial PD are Parkin [16], PTEN-induced putative kinase 1 (PINK1) [17], and Daisuke-Junko-1 (DJ1) [18] genes.
These genes are upstream mutations which appear to increase αS toxicity, suggesting that further advances in understanding αS structure and function may be crucial to understanding and ultimately treating PD.
PD is strongly associated with the appearance of dopaminergic neuronal cytoplasmic inclusions called Lewy bodies. These are the leading pathogenic hallmarks in brain biopsies of PD patients, and are not present in healthy individuals.
In 1997 Lewy body inclusions were shown to contain aggregates of αS [19], a 140 amino acid protein which has consequently been implicated as the likely cause of familial PD [20,21,22]. Further evidence is provided by the fact that duplication, triplication and autosomal dominant missense mutations in the SNCA gene lead to early onset forms of PD. It is now believed that the misfolding and subsequent aggregation of αS is a primary cause of dopaminergic degradation in PD.
This is confounded by a rapidly ageing global population, correlating with an increasing number of sporadic cases of PD. In the United Kingdom alone it is believed that about 0.2% of the population are living with PD, affecting an estimated 127,000 people, and currently costing the NHS approximately £212 million per year [23]. This highlights the importance of discovering new methods to diagnose, treat and especially prevent neurodegeneration associated with PD and related synucleinopathies, and to better understand their pathogenesis.
Effective strategies for preventing or reversing αS aggregation and neurotoxicity are urgently needed to avoid an exponential increase in disease with an ageing population. Recent solid state NMR and cryoEM fibril structures have brought new structural insights to the folding and formation of both native and pathogenic conformations of the αS protein [24,25,26,27].
α-Synuclein: native structure and function
Despite considerable effort, the precise native structure of αS is still poorly defined. It has been variously described as intrinsically disordered [28, 29], helical [30, 31], or a combination of the two [32]. A helix-rich structure has been shown to be more readily populated in the presence of phospholipid membranes [33, 34] (Fig. 1), offering one possible insight towards the functional role of the protein.
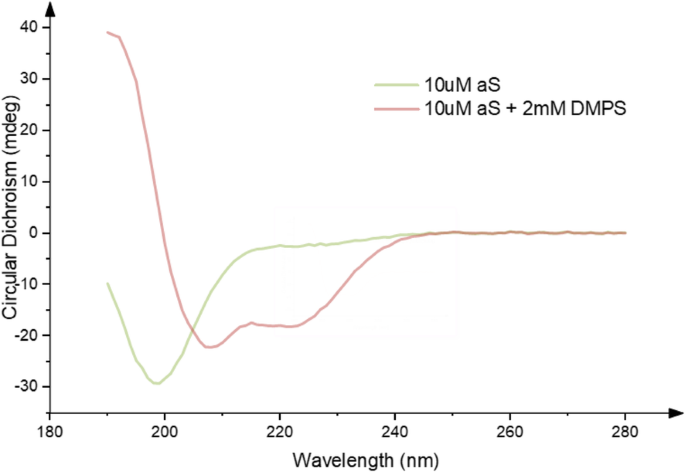
Identifying the precise native state(s) of αS has certainly been hampered by the lack of knowledge of a clear function for the protein, its binding partners, or specific post-translational modifications (see below). The majority of studies have failed to take these variables into account. A wide range of publications have sought to interrogate the structure in a variety of different buffer conditions, including variations in salt, pH and lipid composition [35]. More recently, others have studied different modifications to the protein composition (e.g. phosphorylation, glycation, glycosylation, acetylation) and possible effects on protein structure and function [29, 36, 37].
Some groups have studied protein expression and aggregation in disease-relevant mammalian model systems to identify and understand possible roles for PTMs and the local environment on pathology.
A current consensus is that αS functions to promote membrane curvature, thereby contributing to synaptic trafficking and vesicle budding [38, 39]. This may be important given the association of αS with presynaptic terminal SNARE complexes [40], and suggests a potential role for αS in modulating dopamine release.
This in turn has led to a number of studies investigating the transmission of the protein via synaptic terminals. Additional evidence lends support to a ‘prion-like’ hypothesis, whereby oligomeric αS can migrate between neurons to propagate formation of Lewy bodies throughout the substantia nigra and into extranigral regions.
In particular, Bartels et al [30] and Wang et al [31] independently provided evidence that αS is able to fold into a stable helical structure by associating to form homotetrameric structures. This result was controversial as it was difficult to reproduce in vitro since multimers can disassemble upon cell lysis to generate aggregation prone monomers [41]. Later, others have reported that the structure could be recapitulated by the addition of lipids [42], providing helical multimers and evidence towards a native role for αS association in membrane interactions and in particular, vesicle budding.
A similar effect has been observed either via N-terminal acetylation [43] or by extension of the N-terminus by 10 amino acids [31, 44], leading to formation of a persistent tetramer even in the absence of lipids [30]. Modifications to the N-terminus are known to be particularly important in driving folding towards a helical form of αS [31], which then impacts upon downstream aggregation [45].
Interestingly, a similar homotetrameric model for amyloidogenesis as a general principle had been proposed earlier [46, 47] based on the observed properties of a synthetic homotetramer formed from 4 equivalents of a short Glu/Gln rich peptide deliberately assembled in parallel on an artificial scaffold.
In these experiments the peptide became significantly more α-helical and indefinitely stable at pH 7 when brought together in a parallel alignment, forming a homotetrameric arrangement. However, acidification transformed the α-helical aggregate, via a more elongated 4(310) helix bundle [47] that led to tetramer aggregation, en-route to further elongation into four β-strands, seeding β-sheet aggregation and oligomerisation into matted amyloid-like fibrils.
The key finding was that the tetrameric α-helix bundle was stabilised in water due to its hydrophobic core and polar hydrophilic exterior, like most proteins. However, the α-helix is in equilibrium with its more elongated 310 helix analogue, and transition to a 4(310)-helix bundle proceeds under acidosis conditions due to protonation of hydrophilic residues (Glu). Rearrangement of polar Glu/Gln residues to the interior of the helix core and some hydrophobic residues (Leu) to the exterior surface promotes aggregation.
This led to core destabilization and an α-helix to 4(310)-helix transition driven by inter-coil hydrogen bonds formed between facially paired protonated Glu residues (carboxylic acid dimers) and paired Gln residues (hydrogen bonded carboxamides). These interactions provided the catalyst for driving the equilibrium towards thermodynamically more stable strand/sheet formation and aggregation into oligomeric amyloids.
For that particular peptide sequence, the process could be completely reversed back to the stable α-helical tetramers by restoring the pH to 7. Interestingly, acidosis has been associated with accumulation of αS oligomers [48, 49]. Local acidosis occurs at sites of inflammation and under conditions of metabolic stress (glycolysis and lactic acidosis), but whether this amyloidogenesis model with partial glutamate protonation or interstrand coupling of polar sidechains is relevant to αS oligomerisation and PD is unknown.
The current paradigm is that αS is likely to exist in vivo as an equilibrium mixture of unstructured monomer and statistically disfavoured helical oligomer(s), perhaps partially folded at membranes through phospholipid interactions.
The alpha helical form of the protein may be required for an unknown native function but is not anticipated to be pathogenic, leading to the idea of stabilizing helical αS as a novel intervention strategy for PD.
This might be similar to an approach used by Kelly and co-workers in stabilizing the native transthyretin fold, albeit targeting the protein with small molecules [50].
α-Synuclein Misfolding: implications for PD
Following the implication of the SNCA gene, and therefore αS, as a leading cause of pathology in familial forms of PD (see below) [20,21,22], it was also shown to be the primary protein found within Lewy bodies [19]. In particular, a central hydrophobic region of the protein corresponding to residues 71–82 was found to be essential for the misfolding and aggregation of αS into fibrils. The 71–82 region was also found to be able to aggregate in isolation [51], its deletion (residues 71–82 [51] or 66–74 [52]) preventing aggregation of the protein and implicating these as key regions in misfolding and possibly instigation of amyloidosis. More recently, Tuttle et al. employed ssNMR to demonstrate that the structure of αS in its fibrilar β-sheet arrangement adopts a serpentine Greek key topology [24].
This structure again highlighted the importance of the 71–82 region in stabilizing the pathogenic conformation of αS, but importantly also highlighted a second critical region that is strongly associated with early onset mutations (in particular E46K, H50Q, A53T/E/V and G51D – see below). The region, spanning residues 45–57 is key in mediating β-strand to β-strand interactions in the fibril conformation.
This also reflected an exposed surface on fibrils between residues 46–57, suggesting that this region of αS is accessible in the fibril (see below). More recently, a number of cryoEM structures of mature fibrilar forms of the protein has been solved by two independent research groups [25,26,27, 53] with many similarities to the ssNMR structure.
Two structures display a Greek-key topology, with a further two characterised by a hydrophobic cleft stabilised by intermolecular salt bridges and additional interactions between the NAC and the N-terminus [53] (see below). In all cryoEM structures the fibrils form dimeric strands, with rotational symmetry about the axis. In the former two structure is provided by the seemingly exposed 45–57 region of the fibrillised protein.
This region may therefore act as a hydrophobic ‘steric zipper’, as first described in amyloid fibrils by Eisenberg and colleagues [54], between adjacent protofibrils that then serves to facilitate the formation of a more mature double stranded fibril structure [25, 55].
Genetic evidence for αS in PD
A relationship between genetics and PD was first identified in 1990, when members of an Italian-American family (the Contursi Kindred) were found to manifest inherited early onset PD. Studies subsequently found Lewy body pathology after autopsy [21] and the causative mutation leading to familial early on-set PD was located in the αS gene (SNCA) on chromosome four [20]. The specific mutation was an autosomal-dominant single base pair change in SNCA leading to the A53T substitution in αS [20].
Following this discovery, further autosomal dominant mutations in the SNCA gene have been found to cause familial PD. These include E46K [56,57,58], H50Q [59,60,61,62], G51D [59, 63], A53T [20, 64], A53E [65], A53V [66] and A30P [67,68,69] (Table 1). The most potent of known mutations, leading to the earliest onsets of the disease, is G51D. Interestingly, despite all of these single amino acid changes leading to early onset of PD, each provides very different effects on the αS aggregation rate and the oligomers that become populated.
For instance, the E46K [56,57,58], H50Q [59,60,61,62] and A53T [20, 64] mutations all lead to an increased rate of fibril formation, whereas the G51D [69], A30P [67] and A53E [70] mutations appear to slow the rate of fibril formation. All mutations must therefore lead to either an increase in the aggregation rate, or a change in the oligomeric state or conformation that is populated upon aggregation, as well as a decrease in the normal tetramer:monomer ratios that facilitates these changes.
The mutants collectively provide compelling evidence that aggregation of αS directly leads to early onset PD, while others more specifically provide indirect evidence that prefibrilar oligomers are more toxic than mature aggregated fibrils.
In addition to changes in aggregation kinetics of mutant αS variants, differences in their association with phospholipid membranes have also been observed. Mutations typically result in reduced phospholipid binding, as for example in G51D, A30P [68, 69] and A53E [70] variants. In contrast E46K and A53T lead to increased phospholipid binding [58]. These observations suggest a functional relationship between αS and lipid binding that can become compromised by changes in interaction and structure in early onset mutants.
In addition to missense mutations described above, autosomal dominant familial PD has been observed when the SNCA gene becomes duplicated or triplicated [71, 72]. Consistent with the role of αS in PD, examples where triplication has occurred have led to more severe forms of PD than in instances of gene duplication.
This highlights the importance of intracellular concentrations in driving increased likelihood of αS misfolding, seeding, and ultimately to an early onset of the disease phenotype relative to sporadic cases of PD.
Source:
IOS Press
Media Contacts:
Diana Murray – IOS Press
Image Source:
The image is credited to University of Groningen – The Netherlands.
Original Research: Open access
“Gastrointestinal Immunity and Alpha-Synuclein”. Michael Zasloff et al.
Journal of Parkinson’s Disease doi:10.3233/JPD-191702.



{kind=link}