











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



In suspense novels, the most unassuming character sometimes turns out to be a mastermind, influencing events without attracting attention.
That same storyline may be afoot in cells as well: an unglamorous fat-storing droplet appears to have a surprise role in controlling gene activity.
The droplets prevent cells from activating genes involved in sugar metabolism, Howard Hughes Medical Institute Investigator Tobias Walther and colleagues report February 4, 2020, in the journal Molecular Cell.
“For many years, people thought that fat stored in cells was some sort of enormous glob that just sits there and doesn’t do much,” says Walther, a cell biologist at Harvard University.
His team’s new study topples that idea, establishing seemingly simple fat droplets as key regulators in the complex process of metabolism.

Understanding all the links in that chain of command could help researchers understand the origins of diseases like atherosclerosis, fatty liver disease, or insulin resistance.
Cells use lipids, a kind of fatty molecule, to store energy. Excess fat resides in cellular packets called lipid droplets.
In recent years, scientists have noticed that these droplets interact with molecules involved in metabolism, but the picture remains incomplete.
“Here’s this universal process, and we just don’t understand anything about the genes and the proteins that govern it,” says study coauthor Robert Farese Jr. Walther and Farese, who operate a joint laboratory, set out to identify all the genes coordinating fat storage.
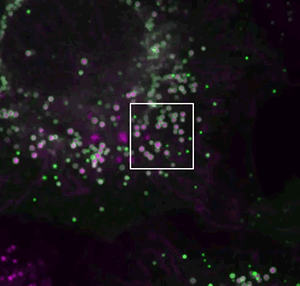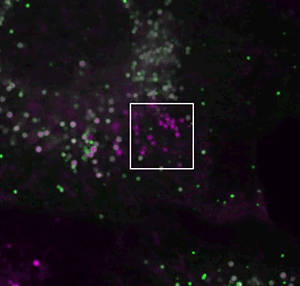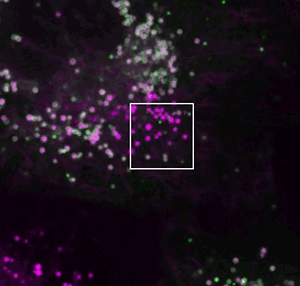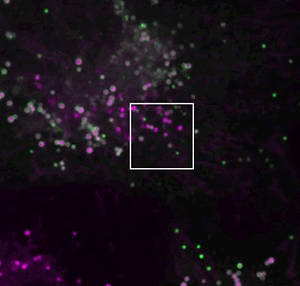
They screened for relevant human genes in white blood cells called macrophages, which store different types of fat. One macrophage gene the team identified surprised them.
The gene, called MLX, encodes a type of protein that generally acts in the nucleus – far away from lipid droplets in the cell.
If MLX influenced lipid metabolism, Walther reasoned, it would probably do so indirectly, by cranking up activity of other genes. But Niklas Mejhert, a postdoc in Walther and Farese’s lab, discovered that MLX could bind to the droplets directly.
That was a startling observation, Walther says. “These proteins work on DNA, so what on earth do they have to do on lipid droplets?”
If MLX is physically stuck to the droplets, the team found, the protein stays trapped outside the nucleus.
There, MLX can’t do its usual job – which includes telling cells to make proteins that burn sugar for energy. Instead, perhaps, cells might burn fat stored in the droplets.
Like a molecular safety valve, the system may prevent cells from becoming overfilled with fat. Too much stored cellular fat can lead to the metabolic diseases that afflict millions of Americans, Farese says.
“Do abnormalities of this system somehow underlie that disease process?”
In addition to MLX and its relatives, the team’s genetic screen surfaced over 500 genes that may participate in fat storage.
“We see that as a virtual treasure trove,” Farese says. The researchers are now working on a website to catalog these genes, so other labs can dive in and explore.
Cells rely on lipids to provide an efficient means of fuel storage and energy production. Lipids are also the major building blocks of cellular membranes and are precursors for hundreds of signaling molecules.
Most eukaryotic cells have the capacity to store lipids in the form of lipid droplets.
Being only recently recognized as dynamic, independent organelles rather than inert fat depots, lipid droplets are now emerging as central regulators of lipid uptake, metabolism, trafficking, and signaling in the cell.
However, their roles extend well beyond lipid metabolism and an increasing number of studies suggest that they play vital roles in the cellular stress response [1–4].
Lipid droplets are dynamically synthesized and broken down in response to cellular needs and environmental signals [5].
Their biogenesis is induced in cells exposed to excess amounts of lipids, to nutrient and oxidative stress, and to various other conditions characterized by energetic and redox imbalances [2].
Intriguingly, lipid droplet biogenesis also occurs in response to complete nutrient deprivation, suggesting that the expense of their synthesis must be worthwhile for the cell and is in fact essential for its response to stress.
Indeed, emerging studies reveal that lipid droplets perform numerous tasks crucial for the protection of cellular integrity and function during stress: they sequester potentially toxic lipids and proteins, maintain energy and redox balance, preserve membrane and organelle homeostasis, modulate autophagy, and provide lipids acting as signaling mediators [1,2,4].
Many of these functions are engaged simultaneously and in cooperation with other organelles and we are only beginning to understand their interplay.
One of the major properties of lipid droplets is their ability to buffer excess lipids, thereby providing immediate protection from lipotoxicity, and to release them later, gradually and according to cellular needs (Figure 1).
This property is particularly important for cells exposed to rapidly changing conditions of stress and it is, for example, exploited by cancer cells during alternating periods of feeding/starvation or hypoxia/reoxygenation [6–10].
This simple aspect of lipid droplet biology – buffering and delayed release of lipids – is indispensable for most of their pleiotropic roles in the cellular stress response.
Lipid droplet buffering and delayed release of lipids. Various conditions of cellular stress are characterized by lipid overload, arising from an excess of exogenous (e.g. increased uptake of lipids from the circulation) or endogenous lipids (e.g. starvation-induced autophagic breakdown of membranous organelles).
Lipid droplets have the capacity to sequester excess lipids, thereby preventing their lipotoxicity, and to release them gradually based on cellular needs to preserve energy, redox, membrane, and organelle homeostasis through various mechanisms.
This essential property of lipid droplets – buffering and delayed release of lipids – is particularly important for cells exposed to rapidly changing conditions of nutrient and oxidative stress.
Lipid Droplets Respond to Nutrient and Energy Imbalances in Stressed Cells
During evolution, cells have developed mechanisms of metabolite sensing in order to cope with the unreliable supply of sugars, amino acids and lipids from the environment [34]. The metabolite-sensing machinery facilitates the coordination of mechanisms that govern nutrient preference, uptake, distribution, recycling, and storage in order to maintain cellular homeostasis.
AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1) are highly conserved and fundamental reciprocal regulators of cellular metabolism [34–37].
AMPK is activated by nutrient and oxidative stress, genotoxins and xenobiotics [37]. It senses minute changes in the intracellular AMP:ATP ratio and responds to low energy states by suppressing anabolic processes, including de novo FA, cholesterol, and TAG synthesis [37–39], while stimulating ATP production through the activation of glycolysis, mitochondrial biogenesis, and FA oxidation [32].
Besides responding to levels of adenine nucleotides, recent reports have shown that AMPK activation may also occur at the lysosomal membrane, where an AMPK-activation complex senses glucose levels independently of the cellular energy status [35,40].
The mTORC1 kinase also localizes to lysosomes, where it is activated by nutrient abundance to promote anabolic cell growth pathways. By regulating the sterol regulatory element-binding protein (SREBP) transcription factors it stimulates FA, cholesterol, and glycerolipid synthesis, whereas it inhibits lipid catabolism by repressing the activity of the transcription factor EB (TFEB) [41–43]. mTORC1 is negatively regulated by AMPK, amino acid, or glucose starvation and is a strong suppressor of autophagy [35,40,44].
Autophagy/lipophagy may be directly regulated by AMPK via a “dual safe” mechanism, involving not only mTORC1 inhibition, but also direct phosphorylation of the Unc-51 like autophagy activating kinase 1 (ULK1) [45].
Different types and severities of nutrient restriction may distinctly affect cellular sensing mechanisms thereby resulting in specific cellular responses. Emerging studies have shown that lipid droplet metabolism is affected by the sensing and regulatory networks of AMPK and mTOR.
In growing yeast cells, lipid precursors are mainly used for membrane synthesis. Following rapid exponential growth, nutrients such as glucose or amino acids are eventually exhausted, which leads to a metabolic shift towards mitochondrial oxidative metabolism.
This is facilitated by the activation of Snf1 (the yeast homologue of AMPK), which senses glucose depletion and derepresses oxidative gene expression [40].
Intriguingly, the switch from fermentation to respiration also includes a rapid burst of lipid droplet biogenesis in order to redirect lipid precursors away from membrane synthesis and preserve them to enable cell survival during the ensuing starvation [3,46,47].
The shift in lipid metabolism is also dependent on the activation of phosphatidate phosphatase 1 (Pah1), which is necessary for TAG synthesis in the stationary phase and is indirectly regulated by TORC1 [48,49].
And how are lipid droplets consumed during starvation?
Yeast cells undergoing sudden glucose restriction consume lipid droplets through microautophagy, i.e. microlipophagy, which is triggered by Snf1/AMPK activation, in order to survive during prolonged nutrient stress [47].
Intriguingly, yeast cells exposed to gradual glucose reduction, amino acid starvation, or TORC1 inhibition cannot activate microlipophagy and do not survive in the long-term, likely because they fail to maintain Snf1/AMPK activation [47].
The mTORC1 kinase may regulate lipid droplet turnover during stress through autophagy-dependent and autophagy-independent mechanisms. In Drosophila, hypoxia-induced TORC1 inhibition leads to autophagy-independent lipid droplet growth and lipid storage in the fat body, which is required for tolerance to hypoxia [50].
In mammalian cells, acute and complete nutrient restriction promotes lipid droplet biogenesis [24]. Experiments in starved mouse embryonic fibroblasts (MEFs) have revealed that amino acid depletion, but not glucose or serum limitation, drives lipid droplet biogenesis [26], which depends on mTORC1 inactivation-induced autophagy [26].
Interestingly, AMPK activation succeeded mTORC1 inhibition and was not required for autophagy initiation, indicating that it might instead be important for sustaining autophagic flux and oxidative metabolism during prolonged starvation [26].
Intriguingly, even in conditions of nutrient sufficiency AMPK may contribute to basal autophagy by activating the ULK1 kinase [51].
Given that lipophagy is activated in milder and prolonged conditions of serum restriction and in the presence of glucose and amino acids [9,25,52], it is not surprising that AMPK stimulates lipophagy through mTORC1-independent activation of ULK1 [51,53].
Thus, it seems that the intricate interplay between mTORC1 and AMPK signaling that regulates autophagy/lipophagy and lipid droplet metabolism depends on the type of nutrients being restricted, the severity and length of stress and the physiological context.
Interestingly, lipolysis and lipophagy are also transcriptionally regulated by common pathways downstream of AMPK and mTORC1 in response to changes in nutrient and energy levels [16,34].
AMPK and mTORC1 signal transduction pathways converge at the TFEB, peroxisome proliferator-activated receptor γ co-activator 1α (PGC1α)-PPARα-retinoid receptor X (RXR) and forkhead box protein O (FOXO) transcription factors that activate the transcription of ATGL and lysosomal acid lipase (LAL), thus promoting both neutral and acid lipolysis [16].
By promoting catabolism and elevating NAD+ levels, AMPK also indirectly activates the deacetylase sirtuin 1 (SIRT1) and its targets PGC1α and FOXO.
On the contrary, starvation-induced mTORC1 inhibition leads to translocation of TFEB into the nucleus, thereby increasing the expression of genes that are involved in lysosomal biogenesis and function, autophagy and lysosomal exocytosis [54].
AMPK also regulates the expression and activation of PLIN3, which promotes lipid droplet dispersion during starvation, and the autophagic degradation of PLIN2, which regulates the binding of cytosolic lipases to the lipid droplet surface [55–58].
At the tissue/organismal level, the role of AMPK in regulating mammalian lipid droplet breakdown is still not clear, with numerous contradictory reports suggesting that AMPK has a pro- or anti-lipolytic function, or has no effect on lipolysis [16].
This suggests a marked tissue- and cell type-specific function of AMPK-mediated regulation of lipid droplet breakdown.
More information: Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Molecular Cell. doi.org/10.1016/j.molcel.2020.01.014



{kind=link}