











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A team of Chinese scientists has recently developed two novel compounds that inhibit the SARS-CoV-2 main protease (Mpro) and one of them is a good drug candidate for further clinical studies.
The research, published online in Science on April 22, was conducted by Prof. LIU Hong and Prof. XU Yechun from the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences (CAS), Prof. YANG Haitao from the Shanghai Institute for Advanced Immunochemical Studies of ShanghaiTech University, Prof. ZHANG Lei-Ke from the Wuhan Institute of Virology of CAS, and their collaborators.
As of April 22, more than 2.5 million cases of COVID-19 have been confirmed, with more than 170 thousand deaths.
No clinically effective vaccines or specific antiviral drugs are currently available for the prevention and treatment of COVID-19.
SARS-CoV-2 – the etiological agent responsible for the global COVID-19 outbreak – is an enveloped, positive-sense, single-stranded RNA virus and SARS-CoV-2 Mpro plays a vital role in its life cycle.
Since SARS-CoV-2 Mpro has no human homologue, it is an ideal antiviral drug target.
After analyzing the substrate-binding pockets of SARS-CoV-2 Mpro, the scientists designed and synthesized two compounds, 11a and 11b.
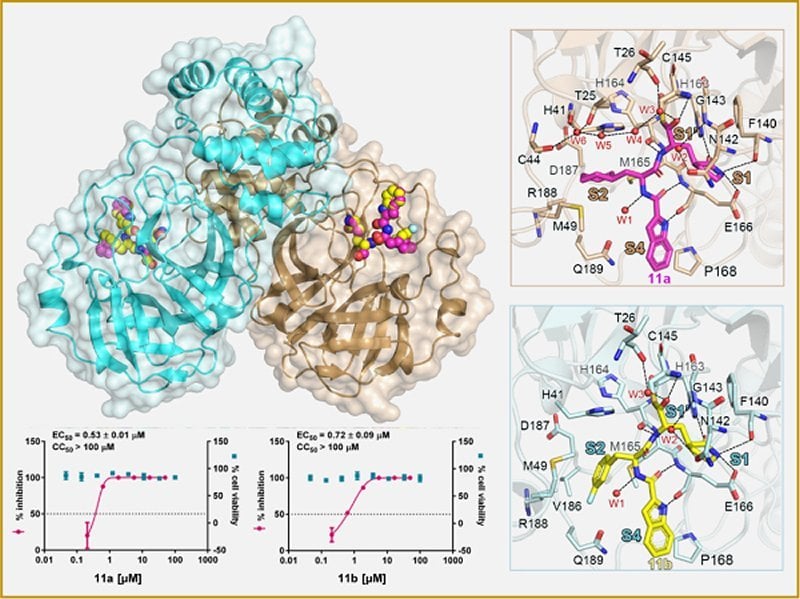
A fluorescence resonance energy transfer (FRET)-based cleavage assay was then used to determine their IC50 values. The results revealed excellent SARS-CoV-2 Mpro inhibitory activity for both 11a and 11b, with IC50 values of 0.053 ± 0.005 μM and 0.040 ± 0.002 μM, respectively.
The researchers also employed immunofluorescence, quantitative real-time PCR and plaque assay to monitor the antiviral activity of 11a and 11b. The results all showed that compounds 11a and 11b exhibited good anti-SARS-CoV-2-infection activity in cell culture (e.g., EC50 values were 0.53 ± 0.01 μM and 0.72 ± 0.09 μM, respectively, when using the plaque assay).
In addition, these compounds showed good PK properties in vivo, suggesting they are promising drug candidates. However, the lower toxicity of compound 11a makes it particularly promising.
In order to elucidate the inhibition mechanism of SARS-CoV-2 Mpro in compounds 11a and 11b, the scientists determined the high-resolution crystal structure of complexes Mpro-11a (PDB: 6LZE) and Mpro-11b (PDB: 6M0K) at 1.5-A resolution.
The high-resolution crystal structure of these complexes not only demonstrated SARS-CoV-2 Mpro-11a/11b interactions, but also revealed the mechanism of SARS-CoV-2 inhibition. High-resolution analysis of complexes is useful to medicinal chemists in designing novel inhibitors that act against SARS-CoV-2.
This study shows that structure-based drug design is an effective strategy for designing specific antiviral leads against SARS-CoV-2. Preclinical research on compound 11a is now proceeding. The team has decided to share its research data with scientists around the world to accelerate the development of anti-SARS-CoV-2 drugs.
In this study, the team led by Profs. LIU Hong, XU Yechun and JIANG Hualiang accomplished the design and synthesis of compounds and evaluated enzyme inhibitory activity; the team led by Profs. YANG Haitao and RAO Zihe determined the high-resolution crystal structure; and the team led by Profs. ZHANG Lei-Ke and XIAO Gengfu tested the antiviral activity. The research was also supported by the National Chengdu Center for Safety Evaluation of Drugs and Frontier Biotechnologies Inc
Comparison of proteases between SARS-CoV-2 and SARS-CoV-1
The SARS-CoV-2 protease consists of three domains (Figure 1A)18 and processes polypro- teins using a catalytic dyad consisting of histidine and cysteine as catalytic residues. Its active site is located between domains I and II. As SARS-CoV-2 is closely related to SARS- CoV-1, their proteases display a high degree of sequence similarity (96.1%; Figure 1D).10,17 In the vicinity of the active site, only a single amino acid (Ser46) is different among the two proteases.
However, the surface topology of the active site among the two proteins presents distinct differences, especially in the vicinity of the loop centered around Asn142 (Figures 1B and 1C). Additionally, the size and depth of the S1’ pocket shows notable differences and, at the center of the S1, S1’, and S3, the SARS-CoV-2 protease presents a more distinct subcavity as opposed to the SARS-CoV-1 enzyme.
Consequently, inhibitors of the SARS protease might present altered binding affinities for the SARS-CoV-2 protease. Similarities to other viral proteases such as the one of HIV or Middle-East Respiratory Syndrome coro- navirus (MERS-CoV) are comparatively low. 22
Although the main protease presumably is the most promising therapeutic target to attenuate viral replication, the inhibition of other functional proteins such as the papain-like protease or the interaction between the viral spike protein and its entry receptor to human cells were considered as well. 8,22

Figure 1: Structural overview and sequence alignment. (A) The three domains (domain I in blue, domain II in grey, domain III in red) of the main protease of SARS-CoV-2 are shown. Amino acid changes between SARS-CoV-1 and SARS-CoV-2 are indicated by asterisks. The cocrystallized ligand (PDB ID 6LU7) is presented in organe. (B) Surface topology of the binding pocket of the SARS-CoV-2 main protease (PDB ID 6LU7). The location of Ser46 is indicated by an asterisk. (C) Surface topology of the binding pocket of the SARS-CoV-1 main protease (PDB ID 2A5I). (D) Sequence alignment of the proteases of SARS-CoV-1 and SARS-CoV-2. Mismatches are marked in blue.
Virtual screening procedures
Virtual Screening is a widely used technique at the early stage of drug discovery that allows to identify potentially bioactive compounds at a high throughput.23 Due to its speed and cost-effectiveness, using it to identify drug candidates against the globally expanding SARS- CoV-2 offers a promising approach, especially when time is of the essence. The inhibition of proteases as treatment against viral infections has been proven for HIV, rendering the SARS-CoV-2 main protease an attractive drug target in particular since its crystal structure has been recently solved. In our virtual screening workflow (Figure 2), we started with 687 million compounds from the three-dimensional ZINC database in a shape-based screening routine. Such a coarse GPU-accelerated protocol allows for the rapid screening of large databases based on known binders as template. Previous virtual screening efforts were limited to significantly smaller compound libraries. 10,12,15,17,20

Figure 2: Virtual screening workflow and binding poses of top two ligands. (A) Virtual screening workflow. (B) Binding pose of CP-1. (C) Binding pose of CP-2.
We refined the high number of initial hits (395132 compounds) to 6599 compounds by selecting the best hits regarding shape overlap. The remaining compounds were docked
into the active site of five representative structures of the protease using the smina dock- ing protocol resulting in 2286 hits with a score below a threshold of -7.0 kcal/mol with the majority of compounds scored below -5.0 kcal/mol. While most previous studies re- garding SARS-CoV-2 were limited to a homology model of the protease,10,12,15,17 we could rely on a recently solved crystal structure for our docking procedures.
The pharmacokinetic descriptors of these compounds are shown in Figure 3. Due to the origin of the selected compounds in the ZINC database, drug-likeness regarding molecular weight (MW) was to be expected. However, several structures violate commonly accepted criteria distribution coefficient logD and polar surface area (PSA), allowing for their delimitation. Due to the catalytic function of the target enzyme, peptides and peptidomimetics are widely applied in targeting proteases.18
Nonetheless, disadvantages of peptides or peptidomimetics include limited oral bioavailability due to their large MW, PSA, and high number of rotatable bonds as well as poor metabolic stability and higher production cost. 24 Therefore, the development of small-molecules with balanced and favorable pharmacokinetic properties facilitating oral absorption offers a promising alternative.

Figure 3: Pharmacokinetically relevant descriptors of the compounds that were subjected to the Glide SP docking protocol.
In the attempt to attain a consensus in binding affinity, we used the Glide SP protocol
to evaluate the interaction of the remaining 2286 compounds with the protein active site. Except for two compounds, a valid binding pose was detected in all cases with most com- pounds scores below -5.0 kcal/mol.
The set of compounds with a Glide score below -6.5 kcal/mol presented a high number of congeneric ligands including many structurally similar natural compounds. Therefore, in order to stimulate the structural diversity of the com- pound set, we computed extended connectivity fingerprints and compared them according to the Tanimoto coefficient for subsequent clustering.
For each cluster, the two compounds with the best Glide score were selected and evaluated regarding their pharmacokinetic de- scriptors. We did not allow the violation of either the Lipinski and Veber criteria resulting in 55 compounds left over for MD simulation. Based on the resulting time-evolved ensemble of ligand-protein complexes, the binding free energies of the ligands were estimated using the MM/GBSA protocol.
This protocol was recently applied to predict the interaction energy between nelfinavir and the SARS-CoV-2 main protease.10
Compounds with a binding free energy improvement in contrast to the cocrystallized ligand were considered regarding their potential toxicity. The VirtualToxLab evaluates the toxic potential of a small molecule on the basis of individual binding affinities to 16 validated off-targets including nuclear receptors, metabolic enzymes, and the human Ether-`a-go-go-Related Gene channel (hERG).
Such an assessment at an early stage of drug discovery might mitigate the attrition rate of drugs due to toxicity and safety which represent a large share of preclinical and clinical failures of drug development programs.25
The remaining 17 compounds included several compounds with a toxic potential above 0.5 that could be discarded from the final set. In the ligands subjected to MD simulation, we further selected the natural compound with the lowest binding free energy which was (-)-taxifolin.
Final compound selection
We identified 11 compounds with a lower free energy of binding combined with a higher theoretical potential of absorption after oral administration compared to the cocrystallized

ligand N3 (Table 1 and Figure 4). The reported set of ligands should be regarded as early lead compounds since no experimentally supported optimization was conducted. While some compounds only offered a minor decrease regarding binding energy, CP-1 to CP-4 presented a substantial improvement in comparison to the cocrystallized ligand up to nearly 30%.
A study that primarily applied artificial intelligence in the form of a neural network to discov- ery SARS-CoV-2 inhibitors additionally tested their compound using AutoDock Vina. Their compounds present docking scores ranging from -7.3 to -8.5 kcal/mol, 12 which is compara- ble to our selection of ligands (Table S1, Supporting Information). Our proposed antivirals interacted with the target with at least one hydrogen bond with an average of over two (Figure 2A and 2B, Table S2).
Further, they showed excellent pharmacokinetic descriptors with logD values ranging from 0 to 4.6, MW below 487 g/mol, PSA below 105.5 ˚A2 and hy- drogen bond acceptors as well as donors in the range described by Lipinski.26 Consequently, the compounds offer an advantage over commonly described peptidomimetic inhibitors. 18
In contrast to other proposed inhibitors for SARS-CoV-2,18 the availability of compounds are marked as either for sale, in-stock or on-demand in the ZINC database (Table S1). Regard- ing potential toxicity through off-target binding, the compounds regularly presented toxic potentials close to 0.5.

The top two compounds CP-1 and CP-2 presented comparatively low values and therefore represent excellent candidates for further investigation in vitro. Sim- ilarly, the natural compound (-)-taxifolin only exhibited a low potential for binding to the 16 off-targets. Natural compounds such as flavonoids were previously considered to inhibit the main protease of SARS-CoV-1 and their effectiveness was demonstrated by fluorescence resonance energy transfer (FRET).18,27
Further, in vivo experiments with (-)-taxifolin evi- denced activity against other viruses such as coxsackievirus B4.28 The sibirian larch (Larix sibirica) is known to produce (-)-taxofolin offering a natural resource for its extraction. Fur- thermore, food preparations containing (-)-taxifolin are readily available from pharmacies and other companies allowing direct and fast access to the potential antiviral.
Remarkably, (-)-taxofolin underwent seven hydrogen bonds which represents the highest count in our se- lection of compounds. Since hydrogen bonds are a key determinant for drug specificity, 29 (-)-taxofolin might offer a naturally occuring alternative to the proposed inhibitors CP-1 to CP-11.
Source:
Chinese Academy of Science
References
(1) Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; Xia, J.; Yu, T.; Zhang, X.; Zhang, L. Epidemiological and clinical character- istics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 2020, 395, 507–513.
(2) Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020,
(3) World Health Organisation, Novel Coronavirus (2019-nCoV) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/ situation-reports/.
(4) Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Scientific Reports 2016, 6, 22677.
(5) Chen, Z.-M. et al. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World journal of pediatrics : WJP
2020,
(6) Wu, F. et al. A new coronavirus associated with human respiratory disease in China.
Nature 2020,
(7) World Health Organisation, International Health Regulations Emer- gency Committee on novel coronavirus in China. https://www. who.int/news-room/events/detail/2020/01/30/default-calendar/
international-health-regulations-emergency-committee-on-novel-coronavirus-in-china.
(8) Stoermer, M. J. Homology Models of the Papain-Like Protease PLpro from Coronavirus 2019-nCoV. 2020,
(9) Keogh-Brown, M. R.; Smith, R. D. The economic impact of SARS: How does the reality match the predictions? Health Policy 2008, 88, 110–120.
(10) Zhijian Xu, Y. S. Z. Z. K. M. X. W. W. Z., Cheng Peng Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. 2020, 1201, 1–20.
(11) Zumla, A.; Hui, D. S.; Azhar, E. I.; Memish, Z. A.; Maeurer, M. Reducing mortality from 2019-nCoV: host-directed therapies should be an option. The Lancet 2020, 395, e35–e36.
(12) Zhang, H.; Saravanan, K. M.; Yang, Y.; Hossain, T. Deep learning based drug screening for novel coronavirus 2019-nCov. 2020, 19, 1–17.
(13) Yang, S. et al. Synthesis, Crystal Structure, Structure-Activity Relationships, and An- tiviral Activity of a Potent SARS Coronavirus 3CL Protease Inhibitor. Journal of Medicinal Chemistry 2006, 49, 4971–4980.
(14) Ghosh, A. K.; Osswald, H. L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. Journal of Medicinal Chemistry 2016, 59, 5172–5208.
(15) Liu, X.; Wang, X.-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. Journal of Genetics and Genomics 2020,
(16) Liu, Z.-B. J. Z. Y. H. R. Z., X. The crystal structure of COVID-19 main protease in complex with an inhibitor N3. http://www.rcsb.org/structure/6LU7.
(17) Li, Y.; Zhang, J.; Wang, N.; Li, H.; Shi, Y.; Guo, G.; Liu, K.; Zeng, H.; Zou, Q. Ther- apeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. bioRxiv 2020, 2020.01.28.922922.
(18) Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S. H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. Journal of Medicinal Chemistry 2016, 59, 6595–6628.
(19) Raugi, D. N.; Smith, R. A.; Gottlieb, G. S. Four Amino Acid Changes in HIV-2 Protease Confer Class-Wide Sensitivity to Protease Inhibitors. Journal of Virology 2016, 90, 1062–1069.
(20) Chen, Y. W.; Yiu, C.-p.; Wong, K.-y. Prediction of the 2019-nCoV 3C-like protease (3CLpro) structure virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates.pdf.crdownload. 2020,
(21) Vedani, A.; Dobler, M.; Hu, Z.; Smieˇsko, M. OpenVirtualToxLab-A platform for gen- erating and exchanging in silico toxicity data. Toxicology Letters 2015, 232, 519–532.
(22) Gao, K.; Nguyen, D. D.; Wang, R.; Wei, G.-W. Machine intelligence design of 2019- nCoV drugs. bioRxiv 2020, 2020.01.30.927889.
(23) Lionta, E.; Spyrou, G.; Vassilatis, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Current Topics in Medicinal Chemistry 2014, 14, 1923–1938.
(24) Recio, C.; Maione, F.; Iqbal, A. J.; Mascolo, N.; De Feo, V. The potential therapeutic application of peptides and peptidomimetics in cardiovascular disease. Frontiers in Pharmacology 2017, 7, 1–11.
(25) Waring, M. J.; Arrowsmith, J.; Leach, A. R.; Leeson, P. D.; Mandrell, S.; Owen, R. M.; Pairaudeau, G.; Pennie, W. D.; Pickett, S. D.; Wang, J.; Wallace, O.; Weir, A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nature Reviews Drug Discovery 2015, 14, 475–486.
(26) Walters, W. P. Going further than Lipinski’s rule in drug design. Expert Opinion on Drug Discovery 2012, 7, 99–107.
(27) Nguyen, T. T. H.; Woo, H. J.; Kang, H. K.; Nguyen, V. D.; Kim, Y. M.; Kim, D. W.; Ahn, S. A.; Xia, Y.; Kim, D. Flavonoid-mediated inhibition of SARS coronavirus 3C- like protease expressed in Pichia pastoris. Biotechnology Letters 2012, 34, 831–838.
(28) Galochkina, A. V.; Anikin, V. B.; Babkin, V. A.; Ostrouhova, L. A.; Zarubaev, V. V. Virus-inhibiting activity of dihydroquercetin, a flavonoid from Larix sibirica, against coxsackievirus B4 in a model of viral pancreatitis. Archives of Virology 2016, 161, 929–938.
(29) Wade, R. C.; Goodford, P. J. The role of hydrogen-bonds in drug binding. Progress in clinical and biological research 1989, 289, 433–444.
(30) Hui, D. S.; I Azhar, E.; Madani, T. A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T. D.; Memish, Z. A.; Drosten, C.; Zumla, A.; Petersen, E. The continu- ing 2019-nCoV epidemic threat of novel coronaviruses to global health — The latest2019 novel coronavirus outbreak in Wuhan, China. International Journal of Infectious Diseases 2020, 91, 264–266.
(31) Schrodinger LCC, Maestro Small-Molecular Drug Discovery Suite 2019-4. 2019,
(32) Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O. G.; Kandrov, D.; Rasputin, K.; Syabro, M.; Tleukenov, T. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167.
(33) Thompson, J. D.; Higgins, D. G.; Gibson, T. J. CLUSTAL W: Improving the sensitiv- ity of progressive multiple sequence alignment through sequence weighting, position- specific gap penalties and weight matrix choice. Nucleic Acids Research 1994, 22, 4673–4680.
(34) Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; Salmon, J.; Shan, Y.; Shaw, D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. ACM/IEEE SC 2006 Conference (SC’06) 2006, 43.
(35) Shaw, D. E. et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. International Conference for High Performance Computing, Networking, Storage and Analysis, SC 2014, 2015- Janua, 41–53.
(36) ChemAxon, Marvin (v.20.4.0). 2020.
(37) Bhal, S. K.; Kassam, K.; Peirson, I. G.; Pearl, G. M. The rule of five revisited: Applying log D in place of log P in drug-likeness filters. Molecular Pharmaceutics 2007, 4, 556– 560.
(38) Sterling, T.; Irwin, J. J. ZINC 15 – Ligand Discovery for Everyone. Journal of Chemical Information and Modeling 2015, 55, 2324–2337.
(39) Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B. A.; Thiessen, P. A.; Yu, B.; Zaslavsky, L.; Zhang, J.; Bolton, E. E. PubChem 2019 update: improved access to chemical data. Nucleic acids research 2019, 47, D1102–D1109.
(40) Koes, D. R.; Baumgartner, M. P.; Camacho, C. J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. Journal of Chemical Informa- tion and Modeling 2013, 53, 1893–1904.
(41) Brooks, B. R. et al. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of computational chemistry 2009, 30, 1545–1614.
(42) Morris, G.; Huey, R. AutoDock4 and AutoDockTools4: Automated docking with selec- tive receptor flexibility. J Comput Chem. 2009, 30, 2785–2791.
(43) Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Pollard, W. T.; Banks, J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. Journal of Medicinal Chemistry 2004, 47, 1750–1759.
(44) Vedani, A.; Dobler, M.; Smieˇsko, M. VirtualToxLab – A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicology and Applied Phar- macology 2012, 261, 142–153.



{kind=link}