










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Coronavirus or COVID-19 has affected many people around the world and is now a major global health threat. COVID-19 was first reported in December 2019 in Wuhan, Hubei Province, China.
In January 2020, the WHO identified it as a new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). According to the WHO report on August 2020, more than 19 million laboratory-approved cases have been reported in 202 countries. Unfortunately, COVID-19 has resulted in more than 700,000 deaths [1].
The major symptoms in infected people include fever, dry cough, aches, pain, tiredness, chills, headache, anorexia, and loss of smell or taste.
Cardiovascular complications may include heart failure, irregular electrical activity in the heart, coagulation disorders, and acute myocardial injury [2]. Moreover, in some people, gastrointestinal symptoms (GI) such as anorexia, nausea, vomiting, diarrhea, and abdominal pain are associated with COVID-19 [3].
These symptoms may start before other symptoms such as fever, aches, and cough.
People infected with COVID-19 also may experience neurological symptoms [4] and these neurological manifestation may occur with or without cardiovascular and respiratory symptoms [5,6]. Specific neurological symptoms accompanying the COVID-19 infection include loss of smell and taste, muscle weakness and pain, tingling in the hands and feet, vertigo, delirium, ischemic and hemorrhagic stroke, and seizures.
Epilepsy is one of the most common, sudden, and recurrent neurological disorders, affecting about 50 million people worldwide. The exact mechanisms leading to seizures are not yet completely understood.
However, the suggested mechanisms include a sever increase in neuronal excitability following an imbalance in the ion channel function, either as an increase in excitatory neurotransmitters of glutamate and aspartate or a decrease in the γ-aminobutyric acid (GABA) neurotransmitter [7].
Other causes of epilepsy include acute metabolic disorders such as hypo or hyperglycemia, electrolyte imbalance, acute neuronal damage following infection and inflammation, stroke, head trauma, mitochondrial dysfunction, hypoxia, and fever.
Only a few studies have been so far conducted to investigate the underlying mechanism of neurological complications of COVID-19, especially seizures and epilepsy. In the following sections, we discuss the five possible mechanisms of epilepsy induced by COVID-19.
COVID-19, Epilepsy and central nervous system inflammation (cytokine storm)
Like all six previous beta-coronaviruses, COVID-19 has the ability to enter the nervous system and causes neurological symptoms. The angiotensin-converting enzyme 2(ACE2) receptor provides the entry route for the coronavirus to infect human host cells.
These receptors are mainly found in the brainstem and are responsible for regulating cardiovascular and respiratory function. Like both the Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS), COVID-19 may also enter the brain directly through the olfactory tract without the need for ACE2 receptors [8].
The neural pathway is a very important way for the virus to enter into the central nervous system. Viruses can travel into the central nervous system either by infecting sensory or motor neurons or by anterograde transport machinery, using kinesin and dynein [9].
After the invasion, the virus triggers reactive astrogliosis and activates the microglia to induce a large inflammatory cascade. Virus entry into the central nervous system leads to the release of pro-inflammatory cytokines (TNF-α, IL-6, IL-1B), nitric oxide, prostaglandin E2, and free radicals, and causes chronic inflammation neural hyper-excitability, seizure, and death [10,11].
Inflammatory cytokines exacerbate apoptosis and neuronal necrosis in the central nervous system, specifically in different parts of the hippocampus, and these pro-inflammatory cytokines play a key role in epileptic pathogenesis. They also cause epilepsy by increasing glutamate and decreasing GABA in the cerebral cortex and hippocampus.
One of the most harmful effects of these cytokines is the secretion of neurotoxic compounds through the autocrine/paracrine mechanisms. These cytokines increase the entry of calcium into neurons through AMPA and NMDA receptors, thereby increasing the neuronal hyper-excitability and death [12,13].
IL-Iβ, which expresses in active microglia and astrocytes, produces the highest concentration of glutamate in the synapses, and increasing the release of glutamate from astrocytes or reducing the reabsorption of glutamate can lead to neuronal hyper-excitability [14].
Laboratory and clinical observations have shown that pro-inflammatory cytokines have a very important role in the onset and maintaining of epilepsy.
IL-1β may also induce seizures by increasing the number of GluN2B subunits in NMDA receptors on post-synapse cells [15,16]. It has been shown that the pathophysiological concentration of IL-1β leads to seizure onset with a resulting decrease in GABA [17].
TNF-α is another pro-inflammatory cytokine released from active microglia and astrocytes. TNF-α increases the release of glutamate from the glia and regulates AMPA receptors [18].
Hyperactive AMPA receptors absorb too much calcium ion and cause neuronal toxicity. Through the endocytosis mechanism, TNF-α not only increases the number of glutamate receptors but also decreases the number of GABA receptors, thereby increasing neuronal excitability [19,20].
IL-6 is the other pro-inflammatory cytokine commonly found in small amounts in a normal central nervous system. However, the stimulation of astrocytes and microglia can lead to an increase in IL-6 production [21].
Other cytokines, such as TNF-α, IL-Iβ, IFN-γ, and IL-17, amplify and increase the production of IL-6 [22]. Studies have revealed that IL-6 reduces long term potentiation (LTP) and neurogenesis of the hippocampus, thereby helping to initiate and increase the severity of epilepsy [23].
Infection of the brainstem with COVID-19 may affect the respiratory and cardiovascular regulatory centers and exacerbate respiratory failure, leading to severe hypoxia. Several data clearly show that acute respiratory distress syndrome and organ failure is the final result of a COVID-19 infection cytokine storm. [24,25].
The combination of hypoxia with pre-existing neuro-inflammation causes severe damage to the hippocampus and cerebral cortex, resulting in neuronal epileptic activity [26, 27, 28, 29].
COVID-19, Epilepsy and BBB breakdown
Activated glia are not the only source for the production of pro-inflammatory cytokines in SARS-CoV-2 infections in the brain. Cytokines, such as IL-6 and TNF-α, can enter the brain through passive or active transmission. Endothelial cells in blood vessels play an important role in the mechanism of blood-brain barrier (BBB) permeability.
COVID-19 infection breaks down the integrity of the BBB, which severely impairs brain homeostasis and leads to neuronal apoptosis and death. On the other hand, the BBB breakdown causes the migration of blood cells and proteins, such as albumin, which disrupt the osmotic balance in central nervous system (CNS) and causes seizure [13,30]. BBB breakdown is the other route of entry of peripheral cytokines to the brain.
The other cause of BBB disruption and seizure-inducement by COVID-19 is fever and hyperthermia. Laboratory studies show that high temperatures (> 40οC) have detrimental effects on various cells, especially metabolic active brain cells, including neurons, microglia, endothelial, and epithelial cells.
Brain damage during extreme hyperthermia increases the acute activation of glial cells and BBB permeability [31]. In children with febrile seizure, fever not only raises the temperature of the brain, but also induces the release of inflammatory mediators, especially cytokines such as interleukin-1β (IL-1β) in the brain. High level of inflammatory cytokines have been detected in cerebrospinal fluid and / or plasma of children with febrile seizure (FS).
COVID-19 may also affect the likelihood of FS. The virus leads to produce inflammatory cytokines in the brains of children which ultimately leads to FS. It has been shown that the expression of IL-1β in reactive astrocytes at least 24 h after FS is increased [32, 33, 34].
COVID-19, abnormal coagulation, stroke and epilepsy
Patients infected with COVID-19 have shown some coagulation abnormalities characterized by prolonged prothrombin time (PT), increased levels of D-dimer, and diffuse intravascular coagulation (DIC). Tang et al. reported that 71.4% of the non-survivors and 0.6% of the survivors of COVID-19 showed evidence of DIC [35, 36, 37].
Several factors may play a role in coagulation disorders in patients with COVID-19. Persistent inflammatory status in COVID-19 patients acts as an important stimulus for a coagulation cascade. Certain cytokines, including IL-6, activates the coagulation cascade and suppresses the fibrinolytic system.
Endothelial damage to the pulmonary and peripheral arteries due to a direct viral attack may be an equally important factor in increasing blood clotting. Endothelial cell damage can activate the coagulation system. Moreover, the immune response can be increased by coagulation disorders.
These two processes may act as a vicious cycle to worsen this situation. In addition, the appearance of antiphospholipid antibodies may impair blood coagulation as well [38, 39, 40, 41].
Post-ischemic and stroke seizure is one of the causes of epilepsy [42]. When a stroke occurs, a seizure may be caused by a variety of factors, including hypoxia, metabolic disorders, and decreased or increased blood perfusion. Acute ischemia may also generate early seizures by increasing extracellular glutamate concentrations, impaired ion channel function, and BBB damage.
The mechanisms involved in late seizures vary and include gliosis, chronic inflammation, angiogenesis, apoptosis and neuronal death, neurogenesis, synaptogenesis, and loss of synaptic plasticity [43,44]. In hemorrhagic stroke, hemosiderin deposits lead to neuronal hyper-excitability and seizures.
The BBB can be broken down by damage to the endothelial cells when serum proteins enter the CNS after a stroke. For example, albumin binds to transforming growth factor beta(TGFβ) receptors in the astrocytes, and TGFβ signaling activates [45]. Subsequently, downregulation of potassium channels Kir4.1 and glutamate transporter occurs.
The result of this event is increasing the potassium and glutamate in the synaptic cleft. An increase in extracellular K leads to seizures. When microglial and astrocytes cells are activated, BBB permeability occurs through the production of pro-inflammatory cytokines such as IL-1β, IL-6, TNFα, and TGFβ. This cycle can amplify epilepsy after a stroke [46, 47, 48].
High levels of glutamate released from ischemic or hypoxic cells into the extracellular spaces may activate AMPA and NMDA receptors leading to neuronal apoptosis or death [49]. GABA is a major neurotransmitter in the nervous system.
Decreased inhibition of this neurotransmitter after stroke leads to excessive neuronal excitability. Animal studies have demonstrated post ischemic encephalopathy in forebrain ischemia which can cause damage to the GABAergic system. The striatum is specifically vulnerable to transient forebrain ischemia.
The dorsolateral striatum has profound neuronal necrosis associated with a marked decrease in GABA synthesis after global ischemia [50]. Decreased GABA receptors may also lead to hyper-excitability of neural networks and seizure [51]. Studies also show that hypoxia, induced by brain ischemia, may play an important role in the onset of epilepsy, depending on how long it lasts. The AMPA receptor antagonist prevents long-term epilepsy after hypoxia [52,53].
COVID-19, mitochondria disturbance and epilepsy
Oxidative stress plays an important role in the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) infection.
Oxidative stress is closely related to mitochondria dysfunction, and the role of mitochondria in the pathology of COVID-19 disease has been confirmed [54, 55, 56, 57].
There is an interplay between mitochondria, oxidative stress, and inflammation during Covid-19 infection. Inflammatory cytokines increase reactive oxygen species (ROS) production in mitochondria [58]. Some inflammatory cytokines, such as TNF-alpha and IL-6, prominent characteristics of the coronavirus found in COVID-19 serum, promote mitochondrial ROS production in the cell.
Mitochondria are intracellular organs with two inner and outer membranes that play an important role in energy homeostasis. In addition to energy production, mitochondria have a variety of functions, including calcium homeostasis, the production of reactive oxygen species (ROS), the modulation of neurotransmitters in the central nervous system, and the regulation of cell apoptosis [59, 60, 61].
There is a reciprocal, cause or consequence relationship between mitochondrial dysfunction and epilepsy. In most types of epilepsy, there is secondary damage to the mitochondria. Mitochondrial dysfunction plays an important role in developing epilepsy.
These organelles are responsible for generating energy in the cells, which is important for the normal electrical activity of neuronal and synaptic transmission. Any disturbance in mitochondrial function may lead to abnormal electrical activity of neurons and produce seizures.
COVID-19, Electrolytes imbalance and epilepsy
Studies have reported various electrolyte abnormalities in patients with coronavirus infection (COVID-19) [62,63]. Electrolyte imbalance may provide insight into the pathophysiology of COVID-19. The COVID-19 infection is associated with decreased serum concentrations of sodium, potassium, magnesium, and calcium, leading to hyponatremia, hypokalemia, hypocalcemia, and hypomagnesemia.
These disorders, especially hypokalemia, may have severe clinical consequences for the infected patient. Hypokalemia leads to exacerbation of ARDS and acute heart damage [10,64,65].
SARS-CoV-2 binds to its host ACE2 receptor, possibly reducing ACE2 expression, thus increasing angiotensin II, which can increase kidneys excretion of potassium, and eventually leads to hypokalemia. Elevated plasma angiotensin II concentrations in patients with COVID-19 act as mediators of acute lung damage, as previously confirmed in SARS-CoV animal models. Potential factors that exacerbate electrolyte imbalance in COVID-19 patients may include gastrointestinal symptoms such as diarrhea and nausea [66, 67, 68].
Seizures are the most important clinical symptoms of electrolyte disturbances and are more common in patients with hyponatremia, hypocalcemia, and hypomagnesemia. In these individuals, successful treatment of seizures begins with an accurate diagnosis of the underlying electrolyte disturbances [69,70].
Early detection and correction of these disorders are essential to control seizures and prevent permanent brain damage. If the electrolyte disorder persists, anti-epileptic drugs (AED) alone is ineffective and inadequate for controlling seizures. The treatment of seizures induced by electrolyte imbalance is determined by the underlying cause and in most cases, AED administration is not necessary until the disturbance is rectified [71, 72, 73].
Conclusion
The impact of the new coronavirus on various organs is not fully understood. Until a definitive and approved cure or vaccine is found, a better understanding of the Covid-19 mechanism leading to organs failure would help to identify strategies and/or therapeutically treatment options for the infection. The virus can cause complicated disorders in the nervous system, such as seizures and epilepsy.
The destructive effects of Covid-19 in the central nervous system are mainly caused by a cytokine storm produced by either the entry of pro-inflammatory cytokines from the periphery into the CNS or the production of these cytokines by activated microglia. Secondary seizures may be initiated after strokes, electrolyte imbalance, increased oxidative stress, and mitochondrial dysfunction in Covid-19 patients. More research is needed to prove the exact mechanism of seizures in Covid-19 patients.
reference link : https://www.msard-journal.com/article/S2211-0348(20)30609-X/fulltext
Neurological Manifestations of COVID-19 Infection
The hallmark of SARS-CoV-2 infection is severe acute respiratory manifestations. However, it was recently documented that, in addition to systemic and respiratory symptoms, some patients may experience neurologic symptoms [2, 11]. Based on a retrospective case series study in Wuhan, China, 36.4% (78/214) of patients with COVID-19 developed neurological symptoms, including headache, nausea or vomiting, fatigue, dizziness, impaired consciousness, acute cerebrovascular problems, ataxia, and seizures [12,13,14].
Hyposmia and hypogeusia are the fairly consistent symptoms of COVID-19 infection [15]. By progress in our knowledge regarding the pathophysiology of COVID-19, it has been suggested that hyposmia could be one of the key early symptoms of COVID-19 infection [16]. Mao and colleagues indicated that severely affected patients are more prone to develop neurological damage than mild or moderate cases.
It must be mentioned that a percent of COVID-19 suffering individuals developed life-threatening conditions, such as acute ischemic stroke (5%), cerebral venous sinus thrombosis (5%), and cerebral hemorrhage (5%) [11]. Postmortem examination of patients with COVID-19 has revealed brain tissue edema and partial neuronal degeneration [17].
Furthermore, the involvement of the central nervous system (CNS) by SARS-CoV-2 has been reported in a subject with viral encephalitis [18]. In keeping with a previous report, a case study claimed that for the first time, they had found an association between frequent seizures and COVID-19 in a young patient. Recurrent seizures could be due to encephalitis and viral invasion to the brain or the toxic effect of inflammatory cytokines [19].
The alteration of angiotensin-converting enzyme 2 (ACE2) receptor expressions might affect the route of virus invasion and pathogenicity of the COVID-19 and may be involved in the transmissibility and severity of disease [20]. Given the various evidence of neurological involvement following SARS-CoV-2 infection, herein, we review the implication of ACE2 in some neurological complications following COVID-19 infection. In the next sections, we focused on the nervous system implications of the current virus.
How Does COVID-19 Invade the Nervous System?
The presence of RNA and protein particles of viruses in the CNS specimens including fluid or parenchyma implies the possibility of a direct invasion of the nervous system by viruses [2].
Indeed, researchers have approved that several viruses (e.g., herpes simplex and herpes zoster) infect either sensory ganglia or motor nerve terminals and then are transmitted to the remote parts of the brain by cellular motor proteins, dynein, and kinesins [2].
Additionally, hemagglutinating encephalomyelitis virus (HEV) and a single-stranded RNA beta coronavirus, in the primary motor cortex of infected rats, have been detected [21]. Moreover, SARS virus genome sequences have been detected in the brain.
According to the clinical and laboratory findings, SARS-CoV-2 (COVID-19) also is a neurotropic virus and can invade nervous tissues, and coating-mediated endo/exocytosis can be a possible route of entry to enter and transmit within the cells [21].
A few theories regarding the way for viral entry into the CNS, particularly the brain, and its pathophysiological mechanisms have been proposed. So far, the most widely accepted theory was that COVID-19 directly attacks the CNS via the olfactory nerve.
It has been shown that CoVs reach the cerebrospinal fluid (CSF) through the olfactory tract a week after the contamination of nasal cells [21]. Interestingly, after the cut of the olfactory bulb, invasion of CoVs into the CNS was sparse in mice [2, 22].
However, based on the current knowledge regarding SARS-CoV-2, direct invasion of the CNS by this virus is controversial, and other routes than the olfactory system should be also considered [23].
ACE2 and angiotensin II (Ang II), the members of the renin-angiotensin system (RAS), are expressed within neurons and astrocytes in different brain regions as well as in the cerebral circumventricular organs and cerebrovascular endothelial cells and play a crucial role in the maintenance of neuroendocrine and autonomic systems, such as the regulation of water and sodium balance, vascular autoregulation, and cerebral blood flow [24, 25].
ACE2 is expressed in different brain structures, particularly in nuclei implicated in the central regulation of cardiovascular function, such as the brainstem, as well as in non-cardiovascular regions, like the motor cortex and raphe [26]. Genetic manipulations in the expression of ACE2 in the whole brain or certain hypothalamic nuclei resulted in a varied pattern of dysfunctions ranging from metabolic and behavioral impairments to disturbed serotonin synthesis and neurogenesis [27].
ACE2 was detected as a functional receptor for the SARS-CoV infection [28]. A growing body of evidence suggests ACE2 as a receptor for spike protein of SARS-CoV-2 [2, 21, 25, 29,30,31]. In addition to ACE2, another proposed target for viral spike protein is CD147 expressing by non-neural cells [32, 33].
Viral spike protein virus may bind to ACE2 and/or CD147 on the host cell and propagate among adjacent cells. CD147, also known as extracellular matrix metalloproteinase inducer, was identified as a red blood cell receptor for the human malaria parasite, Plasmodium falciparum [33].
For this reason, drugs that interfere in the spike protein/CD147 interaction or CD147 expression have been proposed to be beneficial for the control of COVID-19 infection. A reduction of viral load and an improvement of respiratory dysfunction in hospitalized cases have been reported after treatment with azithromycin, presumably via interaction with the CD147 receptor [33].
Given the lack of CD147 in the brain tissue, it likely mediates the CNS damage via indirect mechanisms. However, further findings regarding the effectiveness of azithromycin are controversial [34]. In a study on hospitalized patients with COVID-19, the treatment with hydroxychloroquine and/or azithromycin did not reduce mortality [35].
The more recent report of WHO also confirms that treatment with hydroxychloroquine, another promising drug, does not lower the number of death among hospitalized COVID-19 patients [8]. More evidence is needed to conclude exact antiviral clearance and clinical benefits with the abovementioned drugs in patients with COVID-19 [34, 36].
Although there is rare evidence that CoVs, especially SARS-CoV-2, implicate the nervous system indirectly through the blood circulation pathway [2], whenever the COVID-19 virus reaches the general circulation employing the endothelial or nasal cavity or respiratory epithelial cells and can induce a huge inflammatory response and interrupt the integrity of the blood-brain barrier (BBB).
Next, the virus can pass into the cerebral circulation and viral spike protein interacts with ACE2 expressed in the capillary of cerebral circumventricular organs or interrupted BBB [2]. It has been suggested that viral invasion and penetration occur mainly through ACE2 [2, 22, 25, 27,28,29]. Thus, in the following chapters, we first introduce the RAS and then discuss ACE2 distribution and function in the CNS. Finally, we explain how the RAS axis dysregulation is involved in the pathobiology of SARS-CoV-2.
RAS and Brain
The RAS primarily is an endocrine cascade that regulates blood pressure (BP), vascular wall resistance, and electrolyte balance. Besides its cardiovascular function, this elegant system also plays a significant role in the promotion and maintenance of inflammation and indirectly can affect cognitive function [37].
Renin, a protease secreting by the kidney, catalyzes the first step of the RAS cascade. This enzyme cleaves the angiotensinogen (Agt) secreted by the liver, to produce angiotensin I (Ang I) [38]. Moreover, renin has been recognized within astrocytes and neurons.
Most of the brain Agt is secreted by astrocytes that progress into multiple natural peptides [39]. The ACE converts the Ang I to Ang II, a potent vasoconstrictor, and Ang II stimulates Ang II receptor type1 (AT1R), enhancing BP, inflammation, and even neurodegeneration [38].
Thus, chronic activation of RAS and the increase in Ang II level may activate AT1R, leading to inflammation, fibrosis, and hypertension due to excessive vasoconstriction and increased renal sodium absorption (Fig. 2a). Furthermore, several studies have reported that excessive activation of local RAS in the brain may be implicated in neurodegeneration due to neuroinflammation and excessive oxidative stress.
For example, activation of AT1R by elevated Ang II in the brain tissue perturbs cognitive function, possibly due to increased oxidative stress, neuroinflammation, and cell death. Consequently, the ACE inhibitors (ACEIs) and Ang II receptor blockers (ARBs) improve cognition by reversing these alterations [40]. Both Ang II and AT1R receptors are detectable at nerve terminals of several nuclei in the brain, including the ventrolateral medu
lla, nucleus tractus solitarius, paraventricular nucleus, and subfornical organ (SFO) [39].
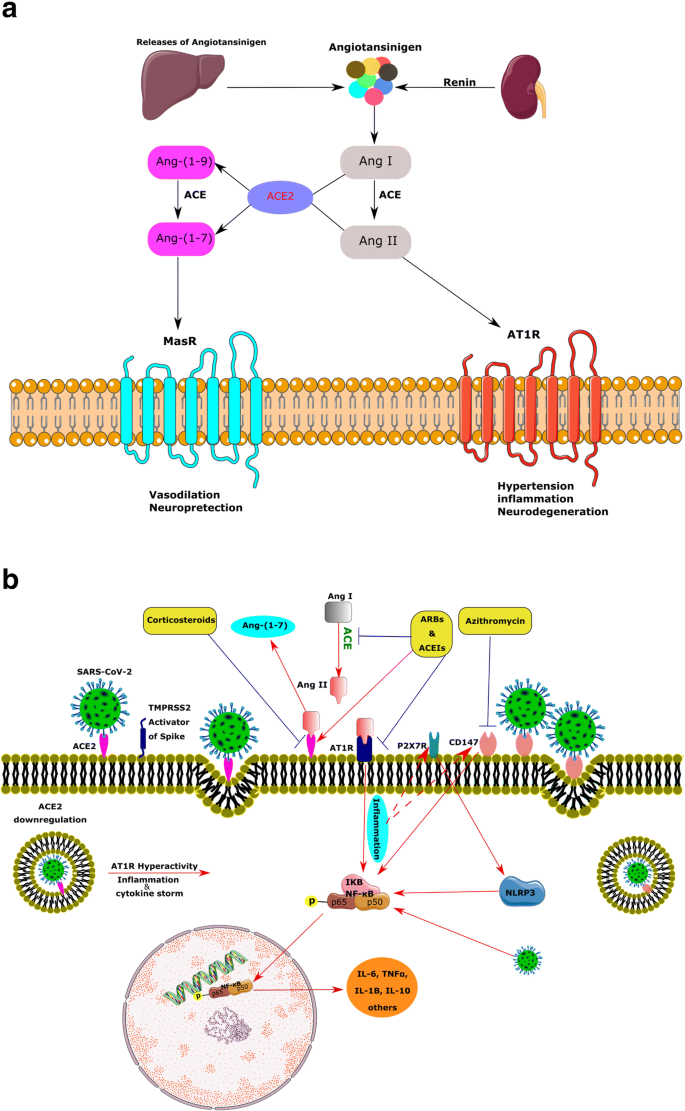
ACE2, a member of the membrane-bound carboxydipeptidase family, is one of the key components and a new member of RAS as well as the regulator of BP [41]. The current enzyme degrades Ang II to Ang-(1–7), an effective 7-amino acid peptide.
Furthermore, ACE2 hydrolyzes the Ang I to Ang-(1–9), and ACE degrades Ang-(1–9) to Ang-(1–7). Ang-(1–7) consequently works upon the Mas receptor (MasR) and has contrasting actions to Ang II, such as vasodilation and neuroprotection. ACE2, Ang-(1–7), and MasR form a new arm for this system (Fig. 2a) [42].
Several investigations support the modulatory role of ACE2 products, such as Ang-(1–7), in the brain. Not surprisingly, expression of Ang-(1–7) is mainly located in nuclei related to BP regulation, such as the brainstem and the hypothalamus, and exerts synergistic or antagonistic effects on Ang-II. Ang-(1–7) has been shown to modulate cardiac baroreflex responsiveness [26].
Overexpression of the abovementioned central RAS members has been shown to elevate the ACE2 protein level in the SFO and affect its expression in the brainstem [43]. Therefore, ACE2 seems to provide a compensatory mechanism to limit brain RAS hyperactivity.
In agreement with this, reducing of AT1R mRNA by a gene silencing approach was associated with a reduction of ACE2 mRNA in the brainstem [44]. In addition, ACE2 participates in the metabolism of some non-RAS peptides, such as hypertensive Apelin-13, neurotensin, and hypotensive bradykinin fragments. Genetic studies have been mapped the ACE2 gene in a hypertension-related quantitative trait locus on the X chromosome. Besides, several studies have shown a strong association of the ACE2 gene polymorphism with hypertension and hypertrophic cardiomyopathy, coronary heart disease, and myocardial infarction [45, 46].
ACE2 Distribution in the Brain
Low levels of ACE2 mRNA have been shown in the human brain using quantitative real-time RT-PCR two decades ago [47]. Furthermore, ACE2 protein was exclusively observed in the endothelial and arterial smooth muscle cells of brain vessels by immunohistochemistry technique [41].
Later, in vitro studies using brain primary cell cultures reported the expression of ACE2 predominantly in the glial cells [48].
In contrast, another study exhibited ACE2 protein and mRNA mostly in the cytoplasm of neuronal cell bodies of mice. Furthermore, profound amounts of ACE2 have also been found in both the central glial substance and in the CSF of human samples [49].
Widespread distribution of ACE2 has been shown throughout the brain, especially in centers involved in the central regulation of cardiovascular function like the brainstem neurons, as well as in non-cardiovascular areas such as the motor cortex and raphe nucleus [50]. In further work, the presence of ACE2 mRNA and protein in the mouse brainstem was also confirmed [44].
According to the data obtained from transgenic mice, ACE2 was significantly high in the SFO. Moreover, a correlation between the ACE2 mRNA and the expression of various proteins in of the nucleus tractus solitaries, the dorsal vagal nucleus, and the caudal ventrolateral medulla has been reported [50], suggesting a crucial role of ACE2 in the modulation of autonomic nervous system.
The expression of ACE2 has been also shown in the olfactory system and other important cerebral regions, such as the brain ventricles and the substantia nigra as well as areas that are directly or indirectly related to the olfactory pathways, including the hypothalamic nuclei, the amygdala, the hippocampus, and the frontal cortex [21].
ACE2 as a Receptor for SARS-CoV-2
In addition to the olfactory system, ACE2 considerably is expressed by epithelial cells of the oral, nasal, and respiratory tract mucosa [51, 52], supporting this idea that ACE2 could be a potential receptor that mediates SARS-CoV2 entry into the cells [2, 23, 53].
As mentioned above, ACE2 has been detected in glial cells and neurons of various brain areas and through the hypothalamus and other nuclei indirectly could make an extra link to the olfactory pathway (Fig. 3a). Nonetheless, how SARS-CoV-2 exactly reaches the CNS through the olfactory pathway is still unknown [23].
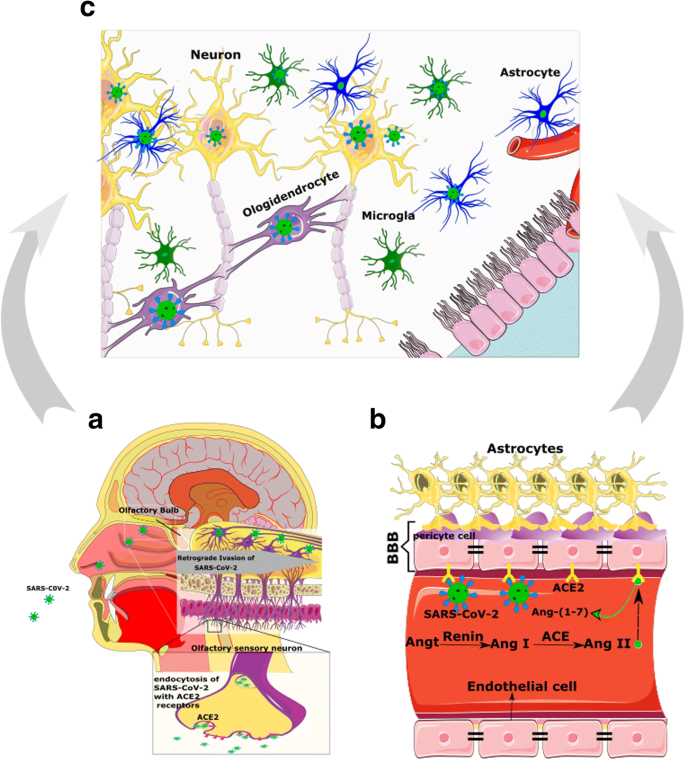
The interaction of viral spike protein and the ACE2 is more complicated than a simple lock-and-key interaction, and other presumed molecules are involved in the process allowing SARS-CoV-2 to invade cells (Fig. 2b). In this scene, Furin and the transmembrane protease serine 2 (TMPRSS2) are two key players [54].
TMPRSS2 locating in the cell membrane primes the spike protein to facilitate viral uptake by ACE2. During the virus/receptor interaction, viral spike protein is cleaved by Furin [55, 56].
Co-expression of ACE2 and TMPRSS2 in epithelial cells but not in sensory neurons of the olfactory system suggests that SARS-CoV-2-induced anosmia, and other olfactory disturbances are related to non-neuronal cells [57]. In contrast to these data, a study using IHC and gene analyses demonstrated that ACE2, TMPRSS2, and Furin are co-expressed in the respiratory and the olfactory mucosa, particularly in the supporting cells of the olfactory epithelium and Bowman’s glands.
This study also has detected ACE2 in the olfactory receptor neurons along with olfactory bulb neurons and concluded that the impairment of the sense of smelling can be due to neuronal dysfunction.
Finally, they declared that direct assault of the olfactory receptor neurons by SARS-CoV-2 is unexpected, while these cells only express ACE2, but not TMPRSS2 or Furin [54]. Therefore, except for direct infection of olfactory neurons, other explanations should be considered for anosmia or hyposmia in patients with COVID-19 (Fig. 3) [21].
Likewise, ACE2 and TMPRSS2 at low levels in human horizontal basal cells (HBCs) have been detected [57]. HBCs are progenitors of the olfactory epithelium and are divided continually to replace sensory neurons during adulthood [58].
Hence, olfactory sensory neurons originated from infected HBCs may be loaded by SARS-CoV-2 and deliver the virus to the olfactory cortex through the olfactory bulb [21]. It must be emphasized that besides the olfactory system, other routes of entry into the CNS should not be neglected [23].
Considering that ACE2 is expressed in the vascular endothelium, pericytes, cerebral neurons, astrocytes, and oligodendrocytes [23, 59], SARS-CoV-2 may break the BBB and invade the CNS by assailing the vascular system. Furthermore, SARS-CoV-2 by binding to ACE2 may raise BP in the brain, enhance the permeability of BBB, and increase the risk of stroke [60, 61] (Fig. 3B and Fig. 4).
Thus, apart from probable direct invasion of neural cells by COVID-19, chronic depletion of brain ACE2 due to occupying by viral particles and perturbing the balance of ACE/ACE2 may also contribute to the neurological manifestations of COVID-19.
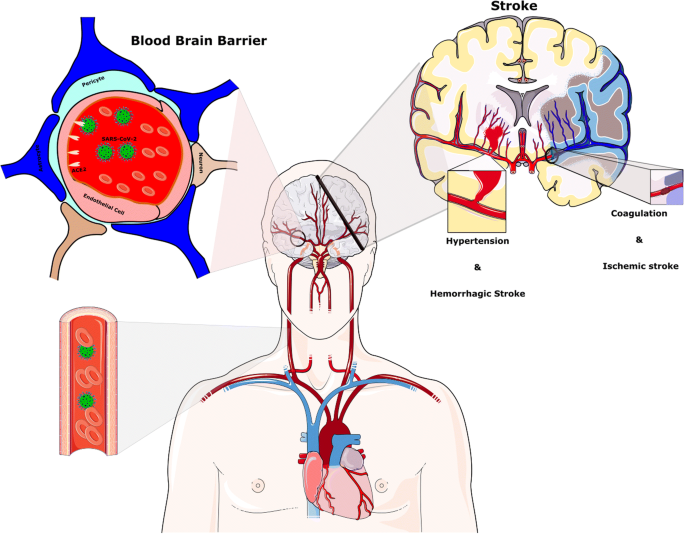
Stroke after the SARS-CoV-2 infection. Hypertension has been generally stated to be associated with SARS-CoV-2 disease. The reduction of ACE2 by SARS-CoV-2 provides the condition for Ang II-dependent hypertension. Raised CRP and D-dimer levels indicate a high inflammatory status and abnormality in the coagulation cascade and probably play a key role in the pathophysiology of stroke in the context of COVID-19 infection
ACE2 and Cytokine Storm, the Main Mechanism of COVID-19 Complications
In the healthy state, the ACE2/MasR axis inhibits the pro-inflammatory actions of ACE/Ang II/AT1R axis by degrading Ang II along with producing Ang-(1–7) and reduces the expression of mitogen-activated protein kinases (MAPK), nuclear factor kappa B (NF-κB), and inflammatory factors, such as interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and IL-8 [62].
On the contrary, hyperactivation of the RAS can induce inflammation due to the release of profibrotic cytokines, such as transforming growth factor-beta (TFG-β), through AT1R/ACE arm. In addition, high levels of Ang II and hyperactivation of AT1R activate the complement cascade, including C5a and C5b-9 [63,64,65].
Under pathologic conditions, such as COVID-19 infection, after binding of viral spike protein to ACE2 in type II pneumocytes, the complex of virus/ACE2 downregulates the intracellular ACE2 and consequently enhances inflammation [62]. As a result of ACE2 downregulation, serum levels of Ang II and the activity of the Ang II/AT1R axis are potentiated [62].
Furthermore, the downregulation of ACE2 leads to the activation of the NF-κB pathway via phosphorylation of its p65 subunit and the enhancement of IL-6, TNFα, IL-1B, and IL-10 production. The MAPK, which plays a critical role in the release of various cytokines, including IL-1, IL-10, IL-12, and TNFα, is also modulated by Ang II [66].
Several clinical studies have indicated a marked increase of pro-inflammatory cytokines, like IL-2, IL-6, and TNF-α, in severe and moderate cases of COVID-19 [67,68,69,70,71,72,73].
Besides, SARS-CoV-2 itself stimulates the NF-κB pathway independent of ACE2 through pattern recognition receptors. Predictably, this surge of cytokines in the serum triggers a cytokine storm that results in acute respiratory distress syndrome (ARDS) in severe cases of COVID-19 [62].
Furthermore, local elevated production of cytokines in the cerebral tissue can induce inflammation [73] and lead to neurologic features observed in COVID-19 patients [74]. It should be also mentioned that viral components are capable to interact with various molecules, such as pattern recognition receptors (e.g., Toll-like receptors), NF-κB, complement cascades, certain transcription factors, and cell apoptosis mediators [62, 75].
Following the entrance of the virus into the CNS, miscellaneous signaling pathways inside or between cells could be modified, and cerebral neurotransmitters and local hormones may be dysregulated.
Referring to the fact that COVID-19-induced ARDS is a consequence of an uncontrolled inflammatory response characterized by cytokines storm, several in vivo and in vitro experiments point to the probable implication of the purinergic ionotropic P2X7 in this process. The P2X7R plays a critical role in the neuroinflammation through its potent stimulatory effects on the NLRP3 inflammasome, which consequently leads to caspase-1 activation, IL-1 β and IL-18 release, and macrophage down-modulation. The SARS-CoV can also directly activate the NLRP3 inflammasome. The pharmacological blockade or genetic knockdown of the P2X7R has substantially reduced inflammatory cell infiltration, cytokine levels, and lung damage. Moreover, in the absence of P2X7R, alveolar macrophage death and pro-IL-1 β release decreased in an in vivo model of lung inflammation. Thus, the P2X7R has been proposed as an inflammatory biomarker for COVID-19 infection [76], which may predict the outcome of neurological complications of patients with COVID-19 infection. The purinergic P2X7 receptor is widely distributed in the brain [77] and is linked to the inflammatory and neurodegenerative changes.
Considering the bold action of hyper-inflammation in the pathophysiology of SARS-CoV-2, non-steroidal anti-inflammatory drugs and corticosteroids may be an effective treatment for alleviating symptoms of COVID-19. Emerging evidence demonstrated the effectiveness of corticosteroids in the control of COVID-19-induced acute damage.
However, the possibility of viral rebound or other side effects in the long-term administration of corticosteroids could not be ruled out [78]. In this regard, a randomized controlled trial reported a higher concentration of SARS-CoV-2 RNA in week 2/3 of infection in those treated with corticosteroids.
Additionally, in a pig model of respiratory CoVs infection, a few doses of dexamethasone in the acute phase of infection diminished acute pro-inflammatory response, but with prolonged administration, there was a risk of viral replication. Nevertheless, some evidence supports the beneficial effects of the limited application of anti-inflammatory drugs in the early stage of COVID-19 infection [78].
Mounting evidence implies the comorbidity of hypertension and other forms of cardiovascular diseases in severe cases of COVID-19 that are commonly treated with ACEIs and ARBs. Theoretically, ACEIs and ARBs are supposed to increase ACE2 level and subsequently the entrance of SARS-CoV-2 into the cells and lead to greater adverse outcomes, such as lung injury. However, according to the experimental studies, ACE2 was protective against lung damages. ACE2 forms anti-inflammatory Ang-(1–7) from Ang II and thus antagonizes the inflammatory action of Ang II.
Moreover, ACEIs via the reduction of formation of Ang II and ARBs through the inhibition of Ang II activity and the blockade of AT1R could control the systemic inflammatory response. By the abovementioned mechanisms, these agents could prevent the development of ARDS, myocarditis, or acute kidney injury, which commonly occur in COVID-19.
Furthermore, ARBs are potential candidates for the treatment of SARS-CoV-2 complications. Taking into account that high level of soluble ACE2 in the serum can block viral spike protein and reduce the uptake of SARS-CoV-2 by ACE2 expressing organs, application of recombinant ACE2 is proposed as a therapeutic approach for COVID-19 [79].
In addition to the ACEIs and AT1R blockers [79], anti-inflammatory drugs, like thiazolidinedione and ibuprofen, can increase the amount of ACE2 [80]. It is not clear whether the poor prognosis of COVID-19 infection in hypertensive and diabetic patients is related to their previous pharmacotherapies and molecular adjustments or due to their ACE2 profile.
According to Fang et al., the vulnerability of an individual to COVID-19 could be a result of both previous therapy and ACE2 gene polymorphisms [80]. Taken as a whole, neither the exact role of hypertension nor the beneficial or the harmful effects of ACEIs or ARBs [79] along with anti-inflammatory agents on the outcomes of COVID-19 have been clarified and must be further explored.
Other Possible Mechanisms
There are several possibilities regarding pathophysiologic mechanisms by which COVID-19 affects brain function. A potential mechanism for neurological mechanisms could be related to SARS-CoV-2-induced hypoxia. Viral invasion disturbs the gas exchange by the respiratory system and leads to general hypoxia. Hypoxia induces anaerobic metabolism in the mitochondria of cerebral cells resulting in overproduction of acid.
High levels of acid cause intracerebral vasodilation, brain edema, obstruction of cerebral blood flow, ischemia, and headache. As a result of continuous hypoxia, intracranial hypertension can appear. In high-risk patients with cardiovascular diseases, hypoxia may also induce the occurrence of acute ischemic stroke and neurologic symptoms (Fig. 4) [2].
Ultimately, it must be mentioned that neurologic complications could be due to secondary infection, not coronavirus itself. While the BBB is impaired because of viral infection, it is easier for other pathogens to reach the CNS. Therefore secondary intracranial infections may cause neurological involvement in patients infected with COVID-19 [2].
Neurologic Manifestations of COVID-19 Infection
After summarizing relevant pathophysiological changes of COVID-19 infection that may lead to neurologic manifestations, we describe the most common neurologic complications observed in the infected individuals.
Hyposmia
One of the common early features of contamination with COVID-19 is hyposmia [81]. The direct connection of the olfactory bulbs with the brain, penetration of COVID-19 into the brain, is expectable. While one possible mechanism is a retrograde transmission of COVID-19, through the olfactory epithelium to the brain [82], other probable routs should not be neglected.
It is remembered that neuronal precursors originating from the subventricular zone (SVZ) migrate and mature through the rostral migratory system until reaching the olfactory bulb. The BBB of SVZ is more penetrable compared with other brain areas due to the lack of pericytes and astrocyte-end feet processes, which envelope the blood vessels [83].
On the other side, SARS-CoV-2 binds to ACE2 and therefore could degrade Ang II to Ang-(1–7) and increase the Ang II level in the bloodstream. It is reported that a high level of angiotensin can evoke apoptotic effects on the neural stem cells [84] and reduce the number of precursors in the SVZ and subsequently diminish the number of migrating cells to the olfactory bulb. As a result of a reduction in the replacement of new neurons in the olfactory bulb, the sense of smell can be disturbed.
Seizure
Another neurologic feature observed in patients with COVID-19 is convulsive seizures [19]. In a study conducted on 70 subjects with MERS-CoV infection, the seizure was reported in 9% of the patients [85]. Additionally, seizures occurred in some patients with COVID-19 [11]. RAS has a vital role in several neurological conditions, including seizures. Consistently inhibition of the renin-angiotensin system prevented seizures in a rat model of epilepsy [86].
Furthermore, the expression of Ang II, AT1R, and AT2R receptors in the hippocampal formation of the patients with temporal lobe epilepsy is upregulated, supporting the potential involvement of the RAS in triggering seizures [87]. The reduction of ACE2 availability, as well as the enhancement of Ang II values and its downstream pro-inflammatory mediators in COVID-19, may contribute to the occurrence of seizures.
Epileptic seizures can enhance the production of cytokines, such as IL-1b and TNFα, which in turn regulate the pathogenesis and course of seizure attacks [10].
Stroke
Up to now, stroke is one of the most serious and fatal adverse outcomes of COVID-19 infection. Several investigations have reported the occurrence of acute cerebrovascular disease, particularly ischemic stroke in patients with severe COVID-19 infection [88,89,90].
These serious complications of COVID-19 are due to thrombolytic events, vascular wall defects, and cerebral hemorrhage. Approximately 6% of severe COVID-19 patients developed the cerebrovascular insult in the course of disease [11]. Avula et al. have reported that four patients with PCR confirmed SARS-CoV-2 infection exhibited a radiographic verification of acute stroke [91].
Moreover, a hospitalized subject for a hidden subtle stroke with right limb weakness with low speech fluency has been diagnosed with COVID-19 [30].
Raised CRP and D-dimer levels, indicating a high inflammatory status and abnormality in the coagulation cascade, likely perform a functional task in the pathophysiology of stroke in the context of COVID-19 infection [53]. The overactivation of the complement cascade in the process of the inflammatory response by SARS-CoV-2 can lead to thrombosis (Fig. 4) [92, 93].
Besides, due to the reduction of ACE2 proteins and a further increase in Ang II, enhanced BP can raise the occurrence of stroke. In agreement with this statement, large vessel occlusion, such as the middle cerebral artery, was correlated with CoVs infection [94]. In addition, an increase in the expression of CD147 in astrocytes has been reported in an experimental stroke model [16].
Ataxia
Acute cerebellar infarction may cause lethargy, dysarthria, and ataxia [95]. Moreover, acute cerebral ataxia may coincide with immune-mediated inflammatory conditions and encephalopathies [96], which are common in the SARS-CoV-2 infection.
Headache
Headache is a common sign of various medical problems, such as viral infection and arterial hypertension. Hypertension-induced headache is usually bilateral and acute and is linked to a sharp increase in diastolic BP ≥ 120 mmHg or systolic BP ≥ 180 mmHg [97,98,99].
The balance between the ACE2 and ACE is important for the regulation of BP [100]. The alteration of the ACE/ACE2 ratio in COVID-19, therefore, may increase BP and trigger headaches. Another plausible cause of headache in COVID-19 sufferers could be the surge of inflammatory cytokines, including TNFα, IL-1, and IL-6 [101, 102]. Intracerebral hypoxia and secondary infection of cerebral tissue are other potential causes of headaches [2].
Encephalopathy
Like the majority of described conditions, encephalopathy is not a specific disease, but a symptom or syndrome accompanied by general brain dysfunction causing by organic or nonorganic roots. Due to the involvement of various organs in the SARS-CoV-2 infection, the risk of encephalopathy is very high [103]. There are several types of encephalopathy, such as hypertensive encephalopathy, hypoxic-ischemic encephalopathy, hepatic encephalopathy, and uremic encephalopathy that may be observed in patients with COVID-19.
Hypertensive Encephalopathy
The hypertensive crisis could have resulted in headaches, seizures, and perturbed consciousness [104]. Hypertensive encephalopathy occurs due to the breakdown of BBB integrity following high BP that leads to cerebral hyper-perfusion and, eventually, brain edema.
In a normal state, the cerebral autoregulatory mechanism protects the BBB from hazardous systolic BP increases [105]. In contrast, remarkably fast accelerations of BP cause orthostatic plasma leakage over the capillaries [106]. Hypertension has been generally stated to be associated with SARS-CoV-2 disease [107].
The reduction of ACE2 by SARS-CoV-2 increases the risk of Ang II-dependent hypertension [108]. This may trigger a cascade that results in the hypertensive encephalopathy through the disruption of BBB integrity and cerebral hyper-perfusion.
Hypoxic-Ischemic Encephalopathy
Hypoxic-ischemic brain injury or encephalopathy (HIE) is a neurovascular and neuro-metabolic syndrome. HIE arises as an outcome of a lack of oxygen or glucose supply or changed cerebral metabolism after severe brain damage.
This encephalopathy results from global hypo-perfusion or hyper-oxygenation rather than from specific vascular infarction in a cerebral area [109]. Some cerebral regions between the anterior and middle cerebral artery may be influenced by HIE, including the basal ganglia, hippocampal formation, cerebellum, and thalamus [110].
Respiratory dysfunction following viral infection could also evoke general hypoxia and therefore induce anaerobic metabolism in the mitochondria of cerebral cells. Low pH grounds intracerebral vasodilation, brain edema, obstruction of cerebral blood flow, and ischemia.
In high-risk patients with cardiovascular diseases, hypoxia may also terminate to acute ischemic stroke and other neurologic symptoms [2, 93]. As mentioned, a common etiology of HIE is cardiovascular injuries, which is mutual among SARA-CoV-2-infected patients [111]. ACE2 is also broadly expressed in coronary endothelial cells, cardiac fibroblasts, and cardiomyocytes [112]. Overexpression of ACE2 exerts a protective function and reverses heart failure, whereas suppression of ACE2 expression can accelerate the progress of heart failure [113].
Hepatic Encephalopathy
Hepatic anomalies and even hepatic failure after infection with COVID-19 have been reported. Hepatic damages could be due to the direct effect of SARS-CoV-2 on the liver or due to the side effects of the medications [114]. Xu and colleagues have reported steatosis and liver injury in a SARS-CoV2 patient [23]. Cardiovascular dysfunction could be another potential cause of COVID-19-related hepatic encephalopathy.
Uremic Encephalopathy
Uremic encephalopathy could be a consequence of acute or chronic renal failure after a fall in the glomerular filtration rate, which is frequently reported in patients with COVID-19. This prompts a syndrome with various symptoms, such as fatigue or seizures [115, 116].
Some studies have reported an amplified incidence of acute renal damage following SARS-CoV-2. It is unknown that this failure is caused by virus-evoked inflammatory reactions or cardiovascular disturbance [117,118,119]. The kidney has an essential role in the control of acid-base balance and also is a crucial regulator of cerebral homeostasis via excretion of toxins and modification of cytokine concentrations. Uremic encephalopathy seemingly occurs following ACE/ACE2 imbalance during COVID-19 infection [120, 121].
Conclusion
RAS and ACE2 play crucial roles in the pathogenesis of neurologic manifestations of COVID-19 infection, presumably through their regulatory effects on blood circulation and BP. Furthermore, ACE may implicate in SARA-CoV-2-related neurologic symptoms via its modulatory effects in the inflammatory and immune responses [119].
According to the growing body of evidence, severe neurologic complications of COVID-19 infection, such as stroke, worsen COVID-19 outcomes. Further studies are warranted to investigate whether pharmacological manipulation of RAS and ACE2 may affect the course and outcomes of COVID-19.
reference link :https://link.springer.com/article/10.1007/s12035-020-02149-0



{kind=link}