











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Il coronavirus o COVID-19 ha colpito molte persone in tutto il mondo ed è ora una delle principali minacce per la salute globale. COVID-19 è stato segnalato per la prima volta nel dicembre 2019 a Wuhan, nella provincia di Hubei, in Cina.
Nel gennaio 2020, l’OMS lo ha identificato come una nuova sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2). Secondo il rapporto dell’OMS dell’agosto 2020, sono stati segnalati oltre 19 milioni di casi approvati in laboratorio in 202 paesi. Sfortunatamente, COVID-19 ha provocato più di 700.000 morti [1].
I principali sintomi nelle persone infette includono febbre, tosse secca, dolori, dolore, stanchezza, brividi, mal di testa, anoressia e perdita dell’olfatto o del gusto.
Le complicanze cardiovascolari possono includere insufficienza cardiaca, attività elettrica irregolare nel cuore, disturbi della coagulazione e danno miocardico acuto [2]. Inoltre, in alcune persone, i sintomi gastrointestinali (GI) come anoressia, nausea, vomito, diarrea e dolore addominale sono associati a COVID-19 [3].
Questi sintomi possono iniziare prima di altri sintomi come febbre, dolori e tosse.
Le persone infettate da COVID-19 possono anche manifestare sintomi neurologici [4] e queste manifestazioni neurologiche possono verificarsi con o senza sintomi cardiovascolari e respiratori [5,6]. Sintomi neurologici specifici che accompagnano l’infezione da COVID-19 includono perdita dell’olfatto e del gusto, debolezza muscolare e dolore, formicolio alle mani e ai piedi, vertigini, delirio, ictus ischemico ed emorragico e convulsioni.
L’epilessia è uno dei disturbi neurologici più comuni, improvvisi e ricorrenti, che colpisce circa 50 milioni di persone in tutto il mondo. I meccanismi esatti che portano alle convulsioni non sono ancora completamente compresi.
Tuttavia, i meccanismi suggeriti includono un grave aumento dell’eccitabilità neuronale a seguito di uno squilibrio nella funzione del canale ionico, sia come aumento dei neurotrasmettitori eccitatori di glutammato e aspartato o come diminuzione del neurotrasmettitore dell’acido γ-amminobutirrico (GABA) [7].
Altre cause di epilessia includono disturbi metabolici acuti come ipo o iperglicemia, squilibrio elettrolitico, danno neuronale acuto a seguito di infezione e infiammazione, ictus, trauma cranico, disfunzione mitocondriale, ipossia e febbre.
Finora sono stati condotti solo pochi studi per indagare il meccanismo alla base delle complicanze neurologiche del COVID-19, in particolare convulsioni ed epilessia. Nelle sezioni seguenti, discutiamo i cinque possibili meccanismi dell’epilessia indotta da COVID-19.
COVID-19, Epilessia e infiammazione del sistema nervoso centrale (tempesta di citochine)
Come tutti e sei i precedenti beta-coronavirus, COVID-19 ha la capacità di entrare nel sistema nervoso e provoca sintomi neurologici. Il recettore dell’enzima di conversione dell’angiotensina 2 (ACE2) fornisce la via di accesso al coronavirus per infettare le cellule ospiti umane.
Questi recettori si trovano principalmente nel tronco cerebrale e sono responsabili della regolazione della funzione cardiovascolare e respiratoria. Come sia la sindrome respiratoria acuta (SARS) che la sindrome respiratoria mediorientale (MERS), anche COVID-19 può entrare nel cervello direttamente attraverso il tratto olfattivo senza la necessità di recettori ACE2 [8].
Il percorso neurale è un modo molto importante per il virus di entrare nel sistema nervoso centrale. I virus possono viaggiare nel sistema nervoso centrale infettando i neuroni sensoriali o motori o mediante macchinari di trasporto anterogrado, utilizzando chinesina e dineina [9].
Dopo l’invasione, il virus innesca l’astrogliosi reattiva e attiva la microglia per indurre una grande cascata infiammatoria. L’ingresso del virus nel sistema nervoso centrale porta al rilascio di citochine pro-infiammatorie (TNF-α, IL-6, IL-1B), ossido nitrico, prostaglandina E2 e radicali liberi e causa infiammazione cronica ipereccitabilità neurale, convulsioni e morte [10,11].
Le citochine infiammatorie esacerbano l’apoptosi e la necrosi neuronale nel sistema nervoso centrale, in particolare in diverse parti dell’ippocampo, e queste citochine pro-infiammatorie svolgono un ruolo chiave nella patogenesi epilettica. Inoltre causano l’epilessia aumentando il glutammato e diminuendo il GABA nella corteccia cerebrale e nell’ippocampo.
Uno degli effetti più dannosi di queste citochine è la secrezione di composti neurotossici attraverso i meccanismi autocrini / paracrini. Queste citochine aumentano l’ingresso di calcio nei neuroni attraverso i recettori AMPA e NMDA, aumentando così l’ipereccitabilità neuronale e la morte [12,13].
IL-Iβ, che si esprime nella microglia attiva e negli astrociti, produce la più alta concentrazione di glutammato nelle sinapsi e l’aumento del rilascio di glutammato dagli astrociti o la riduzione del riassorbimento del glutammato può portare a ipereccitabilità neuronale [14].
Osservazioni di laboratorio e cliniche hanno dimostrato che le citochine pro-infiammatorie hanno un ruolo molto importante nell’insorgenza e nel mantenimento dell’epilessia.
IL-1β può anche indurre convulsioni aumentando il numero di subunità GluN2B nei recettori NMDA sulle cellule post-sinapsi [15,16]. È stato dimostrato che la concentrazione fisiopatologica di IL-1β porta all’insorgenza di convulsioni con conseguente diminuzione del GABA [17].
Il TNF-α è un’altra citochina pro-infiammatoria rilasciata dalla microglia attiva e dagli astrociti. Il TNF-α aumenta il rilascio di glutammato dalla glia e regola i recettori AMPA [18].
I recettori AMPA iperattivi assorbono una quantità eccessiva di ioni calcio e causano tossicità neuronale. Attraverso il meccanismo dell’endocitosi, il TNF-α non solo aumenta il numero di recettori del glutammato ma riduce anche il numero di recettori GABA, aumentando così l’eccitabilità neuronale [19,20].
IL-6 è l’altra citochina pro-infiammatoria comunemente presente in piccole quantità in un normale sistema nervoso centrale. Tuttavia, la stimolazione degli astrociti e della microglia può portare ad un aumento della produzione di IL-6 [21].
Altre citochine, come TNF-α, IL-Iβ, IFN-γ e IL-17, amplificano e aumentano la produzione di IL-6 [22]. Gli studi hanno rivelato che IL-6 riduce il potenziamento a lungo termine (LTP) e la neurogenesi dell’ippocampo, aiutando così a iniziare e ad aumentare la gravità dell’epilessia [23].
L’infezione del tronco cerebrale con COVID-19 può influenzare i centri di regolamentazione respiratoria e cardiovascolare e aggravare l’insufficienza respiratoria, portando a grave ipossia. Diversi dati mostrano chiaramente che la sindrome da distress respiratorio acuto e l’insufficienza d’organo sono il risultato finale di una tempesta di citochine infezione da COVID-19. [24,25].
La combinazione di ipossia con neuroinfiammazione preesistente provoca gravi danni all’ippocampo e alla corteccia cerebrale, con conseguente attività epilettica neuronale [26, 27, 28, 29].
COVID-19, epilessia e ripartizione BBB
Le glia attivate non sono l’unica fonte per la produzione di citochine pro-infiammatorie nelle infezioni da SARS-CoV-2 nel cervello. Le citochine, come IL-6 e TNF-α, possono entrare nel cervello attraverso la trasmissione passiva o attiva. Le cellule endoteliali nei vasi sanguigni svolgono un ruolo importante nel meccanismo di permeabilità della barriera emato-encefalica (BBB).
L’infezione da COVID-19 rompe l’integrità del BBB, che danneggia gravemente l’omeostasi cerebrale e porta all’apoptosi neuronale e alla morte. D’altra parte, la rottura del BBB provoca la migrazione di cellule del sangue e proteine, come l’albumina, che interrompono l’equilibrio osmotico nel sistema nervoso centrale (SNC) e provocano convulsioni [13,30]. La degradazione del BBB è l’altra via di ingresso delle citochine periferiche nel cervello.
L’altra causa di interruzione della BBB e di convulsioni da parte di COVID-19 è la febbre e l’ipertermia . Studi di laboratorio dimostrano che le alte temperature (> 40 ° C) hanno effetti dannosi su varie cellule, in particolare cellule cerebrali attive metaboliche, inclusi neuroni, microglia, cellule endoteliali ed epiteliali.
Il danno cerebrale durante l’ipertermia estrema aumenta l’attivazione acuta delle cellule gliali e la permeabilità BBB [31]. Nei bambini con convulsioni febbrili, la febbre non solo aumenta la temperatura del cervello, ma induce anche il rilascio di mediatori dell’infiammazione, in particolare citochine come l’interleuchina-1β (IL-1β) nel cervello. Un alto livello di citochine infiammatorie è stato rilevato nel liquido cerebrospinale e / o plasma di bambini con convulsioni febbrili (FS).
COVID-19 può anche influenzare la probabilità di FS. Il virus porta a produrre citochine infiammatorie nel cervello dei bambini che alla fine porta a FS. È stato dimostrato che l’espressione di IL-1β negli astrociti reattivi almeno 24 ore dopo la FS è aumentata [32, 33, 34].
COVID-19, coagulazione anormale, ictus ed epilessia
I pazienti infetti da COVID-19 hanno mostrato alcune anomalie della coagulazione caratterizzate da tempo di protrombina prolungato (PT), aumento dei livelli di D-dimero e coagulazione intravascolare diffusa (DIC). Tang et al. ha riferito che il 71,4% dei non sopravvissuti e lo 0,6% dei sopravvissuti a COVID-19 hanno mostrato evidenza di DIC [35, 36, 37].
Diversi fattori possono svolgere un ruolo nei disturbi della coagulazione nei pazienti con COVID-19. Lo stato infiammatorio persistente nei pazienti COVID-19 funge da stimolo importante per una cascata della coagulazione. Alcune citochine, inclusa l’IL-6, attivano la cascata della coagulazione e sopprimono il sistema fibrinolitico.
Il danno endoteliale alle arterie polmonari e periferiche dovuto a un attacco virale diretto può essere un fattore altrettanto importante per aumentare la coagulazione del sangue. Il danno alle cellule endoteliali può attivare il sistema di coagulazione. Inoltre, la risposta immunitaria può essere aumentata dai disturbi della coagulazione.
Questi due processi possono agire come un circolo vizioso per peggiorare questa situazione. Inoltre, la comparsa di anticorpi antifosfolipidi può anche alterare la coagulazione del sangue [38, 39, 40, 41].
Le crisi post-ischemiche e l’ictus sono una delle cause dell’epilessia [42]. Quando si verifica un ictus, una crisi può essere causata da una varietà di fattori, tra cui ipossia, disturbi metabolici e diminuzione o aumento della perfusione sanguigna. L’ischemia acuta può anche generare convulsioni precoci aumentando le concentrazioni di glutammato extracellulare, alterata funzione del canale ionico e danno BBB.
I meccanismi coinvolti nelle crisi tardive variano e comprendono gliosi, infiammazione cronica, angiogenesi, apoptosi e morte neuronale, neurogenesi, sinaptogenesi e perdita di plasticità sinaptica [43,44]. Nell’ictus emorragico, i depositi di emosiderina portano a ipereccitabilità neuronale e convulsioni.
Il BBB può essere scomposto da danni alle cellule endoteliali quando le proteine del siero entrano nel sistema nervoso centrale dopo un ictus. Ad esempio, l’albumina si lega ai recettori del fattore di crescita beta trasformante (TGFβ) negli astrociti e la segnalazione del TGFβ si attiva [45]. Successivamente, si verifica una sottoregolazione dei canali del potassio Kir4.1 e del trasportatore del glutammato.
Il risultato di questo evento è l’aumento del potassio e del glutammato nella fessura sinaptica. Un aumento del K extracellulare porta a convulsioni. Quando le cellule della microglia e degli astrociti vengono attivate, la permeabilità al BBB avviene attraverso la produzione di citochine pro-infiammatorie come IL-1β, IL-6, TNFα e TGFβ. Questo ciclo può amplificare l’epilessia dopo un ictus [46, 47, 48].
Alti livelli di glutammato rilasciato da cellule ischemiche o ipossiche negli spazi extracellulari possono attivare i recettori AMPA e NMDA che portano all’apoptosi neuronale o alla morte [49]. GABA è uno dei principali neurotrasmettitori del sistema nervoso.
La diminuzione dell’inibizione di questo neurotrasmettitore dopo l’ictus porta a un’eccitabilità neuronale eccessiva. Studi sugli animali hanno dimostrato l’encefalopatia post ischemica nell’ischemia del proencefalo che può causare danni al sistema GABAergico. Lo striato è particolarmente vulnerabile all’ischemia transitoria del proencefalo.
Lo striato dorsolaterale ha una profonda necrosi neuronale associata a una marcata diminuzione della sintesi di GABA dopo ischemia globale [50]. La diminuzione dei recettori GABA può anche portare a ipereccitabilità delle reti neurali e convulsioni [51]. Gli studi dimostrano anche che l’ipossia, indotta dall’ischemia cerebrale, può svolgere un ruolo importante nell’insorgenza dell’epilessia, a seconda della sua durata. L’antagonista del recettore AMPA previene l’epilessia a lungo termine dopo l’ipossia [52,53].
COVID-19, disturbi dei mitocondri ed epilessia
Lo stress ossidativo gioca un ruolo importante nell’infezione da Coronavirus della sindrome respiratoria acuta grave (SARS-CoV).
Lo stress ossidativo è strettamente correlato alla disfunzione dei mitocondri ed è stato confermato il ruolo dei mitocondri nella patologia della malattia COVID-19 [54, 55, 56, 57].
Esiste un’interazione tra mitocondri, stress ossidativo e infiammazione durante l’infezione da Covid-19. Le citochine infiammatorie aumentano la produzione di specie reattive dell’ossigeno (ROS) nei mitocondri [58]. Alcune citochine infiammatorie, come TNF-alfa e IL-6, caratteristiche prominenti del coronavirus presenti nel siero COVID-19, promuovono la produzione di ROS mitocondriali nella cellula.
I mitocondri sono organi intracellulari con due membrane interne ed esterne che svolgono un ruolo importante nell’omeostasi energetica. Oltre alla produzione di energia, i mitocondri hanno una varietà di funzioni, tra cui l’omeostasi del calcio, la produzione di specie reattive dell’ossigeno (ROS), la modulazione dei neurotrasmettitori nel sistema nervoso centrale e la regolazione dell’apoptosi cellulare [59, 60, 61] .
Esiste una relazione reciproca, causa o conseguenza tra disfunzione mitocondriale ed epilessia. Nella maggior parte dei tipi di epilessia, c’è un danno secondario ai mitocondri. La disfunzione mitocondriale gioca un ruolo importante nello sviluppo dell’epilessia.
Questi organelli sono responsabili della generazione di energia nelle cellule, che è importante per la normale attività elettrica della trasmissione neuronale e sinaptica. Qualsiasi disturbo nella funzione mitocondriale può portare ad un’attività elettrica anormale dei neuroni e produrre convulsioni.
COVID-19, Squilibrio elettrolitico ed epilessia
Gli studi hanno riportato varie anomalie elettrolitiche in pazienti con infezione da coronavirus (COVID-19) [62,63]. Lo squilibrio elettrolitico può fornire informazioni sulla fisiopatologia di COVID-19. L’infezione da COVID-19 è associata a concentrazioni sieriche ridotte di sodio, potassio, magnesio e calcio, che portano a iponatriemia, ipopotassiemia, ipocalcemia e ipomagnesemia.
Questi disturbi, in particolare l’ipopotassiemia, possono avere gravi conseguenze cliniche per il paziente infetto. L’ipopotassiemia porta all’esacerbazione dell’ARDS e al danno cardiaco acuto [10,64,65].
SARS-CoV-2 si lega al suo recettore ACE2 ospite, riducendo eventualmente l’espressione di ACE2, aumentando così l’angiotensina II, che può aumentare l’escrezione renale di potassio e alla fine porta a ipopotassiemia. Elevate concentrazioni plasmatiche di angiotensina II nei pazienti con COVID-19 agiscono come mediatori del danno polmonare acuto, come precedentemente confermato nei modelli animali SARS-CoV. I potenziali fattori che esacerbano lo squilibrio elettrolitico nei pazienti COVID-19 possono includere sintomi gastrointestinali come diarrea e nausea [66, 67, 68].
Le convulsioni sono i sintomi clinici più importanti dei disturbi elettrolitici e sono più comuni nei pazienti con iponatriemia, ipocalcemia e ipomagnesiemia. In questi individui, il trattamento efficace delle crisi inizia con una diagnosi accurata dei disturbi elettrolitici sottostanti [69,70].
La diagnosi precoce e la correzione di questi disturbi sono essenziali per controllare le convulsioni e prevenire danni permanenti al cervello. Se il disturbo elettrolitico persiste, i farmaci antiepilettici (DAE) da soli sono inefficaci e inadeguati per il controllo delle crisi. Il trattamento delle convulsioni indotte dallo squilibrio elettrolitico è determinato dalla causa sottostante e nella maggior parte dei casi la somministrazione di DAE non è necessaria fino a quando il disturbo non viene corretto [71, 72, 73].
Conclusione
L’impatto del nuovo coronavirus su vari organi non è completamente compreso. Fino a quando non si troverà una cura o un vaccino definitivo e approvato, una migliore comprensione del meccanismo Covid-19 che porta a insufficienza degli organi aiuterebbe a identificare strategie e / o opzioni di trattamento terapeutico per l’infezione. Il virus può causare disturbi complicati nel sistema nervoso, come convulsioni ed epilessia.
Gli effetti distruttivi del Covid-19 nel sistema nervoso centrale sono principalmente causati da una tempesta di citochine prodotta dall’ingresso di citochine pro-infiammatorie dalla periferia nel SNC o dalla produzione di queste citochine da parte della microglia attivata. Le crisi secondarie possono essere iniziate dopo ictus, squilibrio elettrolitico, aumento dello stress ossidativo e disfunzione mitocondriale nei pazienti con Covid-19. Sono necessarie ulteriori ricerche per dimostrare l’esatto meccanismo delle convulsioni nei pazienti con Covid-19.
link di riferimento: https://www.msard-journal.com/article/S2211-0348(20)30609-X/fulltext
Manifestazioni neurologiche dell’infezione da COVID-19
Il segno distintivo dell’infezione da SARS-CoV-2 sono le manifestazioni respiratorie acute gravi. Tuttavia, è stato recentemente documentato che, oltre ai sintomi sistemici e respiratori, alcuni pazienti possono manifestare sintomi neurologici [2, 11]. Sulla base di uno studio retrospettivo di una serie di casi a Wuhan, in Cina, il 36,4% (78/214) dei pazienti con COVID-19 ha sviluppato sintomi neurologici, inclusi mal di testa, nausea o vomito, affaticamento, vertigini, alterazione della coscienza, problemi cerebrovascolari acuti, atassia e convulsioni [12,13,14].
Iposmia e ipogeusia sono i sintomi abbastanza coerenti dell’infezione da COVID-19 [15]. Dal progresso della nostra conoscenza riguardo alla fisiopatologia del COVID-19, è stato suggerito che l’iposmia potrebbe essere uno dei primi sintomi chiave dell’infezione da COVID-19 [16]. Mao e colleghi hanno indicato che i pazienti gravemente colpiti sono più inclini a sviluppare danni neurologici rispetto ai casi lievi o moderati.
Va detto che una percentuale degli individui affetti da COVID-19 ha sviluppato condizioni pericolose per la vita, come ictus ischemico acuto (5%), trombosi del seno venoso cerebrale (5%) ed emorragia cerebrale (5%) [11]. L’esame post-mortem dei pazienti con COVID-19 ha rivelato edema del tessuto cerebrale e degenerazione neuronale parziale [17].
Inoltre, il coinvolgimento del sistema nervoso centrale (SNC) da parte di SARS-CoV-2 è stato segnalato in un soggetto con encefalite virale [18]. In linea con un rapporto precedente, un caso di studio affermava che per la prima volta avevano trovato un’associazione tra convulsioni frequenti e COVID-19 in un giovane paziente. Le convulsioni ricorrenti potrebbero essere dovute a encefalite e invasione virale nel cervello o all’effetto tossico delle citochine infiammatorie [19].
L’alterazione delle espressioni del recettore dell’enzima di conversione dell’angiotensina 2 (ACE2) potrebbe influenzare la via di invasione del virus e la patogenicità del COVID-19 e potrebbe essere coinvolta nella trasmissibilità e gravità della malattia [20]. Date le varie prove di coinvolgimento neurologico a seguito di infezione da SARS-CoV-2, qui esaminiamo l’implicazione di ACE2 in alcune complicazioni neurologiche a seguito di infezione da COVID-19. Nelle sezioni successive, ci siamo concentrati sulle implicazioni sul sistema nervoso del virus attuale.
In che modo COVID-19 invade il sistema nervoso?
La presenza di RNA e particelle proteiche di virus nei campioni del SNC, inclusi liquidi o parenchima, implica la possibilità di un’invasione diretta del sistema nervoso da parte dei virus [2].
Infatti, i ricercatori hanno approvato che diversi virus (ad esempio, herpes simplex e herpes zoster) infettano i gangli sensoriali o le terminazioni nervose motorie e quindi vengono trasmessi alle parti remote del cervello da proteine motorie cellulari, dineina e chinesine [2].
Inoltre, sono stati rilevati il virus dell’encefalomielite emoagglutinante (HEV) e un RNA beta coronavirus a filamento singolo, nella corteccia motoria primaria dei ratti infetti [21]. Inoltre, le sequenze del genoma del virus della SARS sono state rilevate nel cervello.
Secondo i risultati clinici e di laboratorio, SARS-CoV-2 (COVID-19) è anche un virus neurotropico e può invadere i tessuti nervosi e l’endo / esocitosi mediata dal rivestimento può essere una possibile via di ingresso per entrare e trasmettere all’interno delle cellule [21].
Sono state proposte alcune teorie sulla via dell’ingresso virale nel SNC, in particolare nel cervello, e sui suoi meccanismi fisiopatologici. Finora, la teoria più ampiamente accettata era che COVID-19 attacca direttamente il sistema nervoso centrale tramite il nervo olfattivo.
È stato dimostrato che i CoV raggiungono il liquido cerebrospinale (CSF) attraverso il tratto olfattivo una settimana dopo la contaminazione delle cellule nasali [21]. È interessante notare che, dopo il taglio del bulbo olfattivo, l’invasione di CoV nel sistema nervoso centrale era scarsa nei topi [2, 22].
Tuttavia, sulla base delle attuali conoscenze riguardanti SARS-CoV-2, l’invasione diretta del sistema nervoso centrale da parte di questo virus è controversa e dovrebbero essere considerate anche altre vie oltre al sistema olfattivo [23].
L’ACE2 e l’angiotensina II (Ang II), i membri del sistema renina-angiotensina (RAS), sono espressi nei neuroni e negli astrociti in diverse regioni del cervello, negli organi cerebrali circumventricolari e nelle cellule endoteliali cerebrovascolari e svolgono un ruolo cruciale nel mantenimento dei sistemi neuroendocrino e autonomo, come la regolazione dell’equilibrio idrico e del sodio, l’autoregolazione vascolare e il flusso sanguigno cerebrale [24, 25].
L’ACE2 è espresso in diverse strutture cerebrali, in particolare nei nuclei implicati nella regolazione centrale della funzione cardiovascolare, come il tronco encefalico, così come nelle regioni non cardiovascolari, come la corteccia motoria e il rafe [26]. Le manipolazioni genetiche nell’espressione di ACE2 nell’intero cervello o in alcuni nuclei ipotalamici hanno prodotto un modello vario di disfunzioni che vanno da disturbi metabolici e comportamentali a disturbi della sintesi e neurogenesi della serotonina [27].
ACE2 è stato rilevato come recettore funzionale per l’infezione da SARS-CoV [28]. Un numero crescente di prove suggerisce che l’ACE2 sia un recettore per la proteina spike di SARS-CoV-2 [2, 21, 25, 29,30,31]. Oltre ad ACE2, un altro bersaglio proposto per la proteina spike virale è il CD147 espresso da cellule non neurali [32, 33].
Il virus della proteina spike virale può legarsi ad ACE2 e / o CD147 sulla cellula ospite e propagarsi tra le cellule adiacenti. Il CD147, noto anche come induttore della metalloproteinasi della matrice extracellulare, è stato identificato come un recettore dei globuli rossi per il parassita della malaria umana, Plasmodium falciparum [33].
Per questo motivo, è stato proposto che i farmaci che interferiscono nell’interazione proteina spike / CD147 o nell’espressione CD147 siano utili per il controllo dell’infezione da COVID-19. Dopo il trattamento con azitromicina sono stati riportati una riduzione della carica virale e un miglioramento della disfunzione respiratoria nei casi ospedalizzati, presumibilmente tramite l’interazione con il recettore CD147 [33].
Data la mancanza di CD147 nel tessuto cerebrale, probabilmente media il danno al SNC tramite meccanismi indiretti. Tuttavia, ulteriori risultati riguardanti l’efficacia dell’azitromicina sono controversi [34]. In uno studio su pazienti ospedalizzati con COVID-19, il trattamento con idrossiclorochina e / o azitromicina non ha ridotto la mortalità [35].
Il rapporto più recente dell’OMS conferma anche che il trattamento con idrossiclorochina, un altro farmaco promettente, non riduce il numero di decessi tra i pazienti COVID-19 ospedalizzati [8]. Sono necessarie ulteriori prove per concludere l’esatta clearance antivirale e benefici clinici con i suddetti farmaci nei pazienti con COVID-19 [34, 36].
Sebbene vi siano rare prove che i CoV, in particolare SARS-CoV-2, implichino il sistema nervoso indirettamente attraverso la via della circolazione sanguigna [2], ogni volta che il virus COVID-19 raggiunge la circolazione generale impiegando la cavità endoteliale o nasale o le cellule epiteliali respiratorie e può indurre un’enorme risposta infiammatoria e interrompere l’integrità della barriera emato-encefalica (BBB).
Successivamente, il virus può passare nella circolazione cerebrale e la proteina spike virale interagisce con ACE2 espresso nel capillare degli organi cerebrali circumventricolari o BBB interrotto [2]. È stato suggerito che l’invasione e la penetrazione virale avvengano principalmente attraverso ACE2 [2, 22, 25, 27,28,29]. Pertanto, nei capitoli seguenti, introdurremo prima il RAS e poi discuteremo la distribuzione e la funzione di ACE2 nel CNS. Infine, spieghiamo come la disregolazione dell’asse RAS è coinvolta nella patobiologia di SARS-CoV-2.
RAS e cervello
Il RAS è principalmente una cascata endocrina che regola la pressione sanguigna (PA), la resistenza della parete vascolare e l’equilibrio elettrolitico. Oltre alla sua funzione cardiovascolare, questo elegante sistema svolge anche un ruolo significativo nella promozione e nel mantenimento dell’infiammazione e indirettamente può influenzare la funzione cognitiva [37].
La renina, una proteasi che secerne dal rene, catalizza il primo passo della cascata RAS. Questo enzima scinde l’angiotensinogeno (Agt) secreto dal fegato, per produrre angiotensina I (Ang I) [38]. Inoltre, la renina è stata riconosciuta negli astrociti e nei neuroni.
La maggior parte del cervello Agt è secreto dagli astrociti che progrediscono in più peptidi naturali [39]. L’ACE converte l’Ang I in Ang II, un potente vasocostrittore, e Ang II stimola il recettore Ang II tipo1 (AT1R), aumentando la pressione arteriosa, l’infiammazione e persino la neurodegenerazione [38].
Pertanto, l’attivazione cronica di RAS e l’aumento del livello di Ang II possono attivare AT1R, portando a infiammazione, fibrosi e ipertensione a causa dell’eccessiva vasocostrizione e dell’aumentato assorbimento renale del sodio (Fig. 2a). Inoltre, diversi studi hanno riportato che un’eccessiva attivazione della RAS locale nel cervello può essere implicata nella neurodegenerazione a causa della neuroinfiammazione e dell’eccessivo stress ossidativo.
Ad esempio, l’attivazione di AT1R da parte di un Ang II elevato nel tessuto cerebrale perturba la funzione cognitiva, probabilmente a causa di un aumento dello stress ossidativo, della neuroinfiammazione e della morte cellulare. Di conseguenza, gli ACE-inibitori (ACEI) e i bloccanti del recettore Ang II (ARB) migliorano la cognizione invertendo queste alterazioni [40]. Entrambi i recettori Ang II e AT1R sono rilevabili ai terminali nervosi di diversi nuclei del cervello, compreso il medu ventrolaterale
la, nucleo del tratto solitario, nucleo paraventricolare e organo subfornicale (SFO) [39].
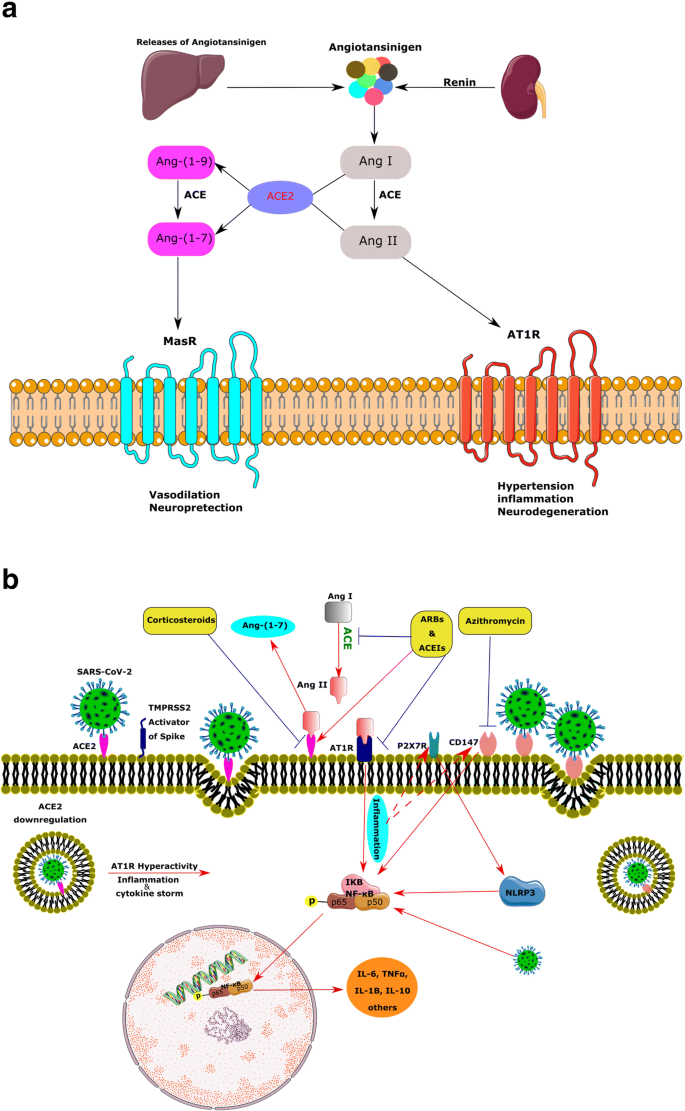
L’ACE2, un membro della famiglia delle carbossidipeptidasi legate alla membrana, è uno dei componenti chiave e un nuovo membro del RAS, nonché il regolatore della BP [41]. L’attuale enzima degrada Ang II in Ang- (1–7), un efficace peptide di 7 aminoacidi.
Inoltre, ACE2 idrolizza Ang I in Ang- (1–9) e ACE degrada Ang- (1–9) in Ang- (1–7). Ang- (1–7) di conseguenza agisce sul recettore Mas (MasR) e ha azioni contrastanti con Ang II, come vasodilatazione e neuroprotezione. ACE2, Ang- (1–7) e MasR formano un nuovo braccio per questo sistema (Fig. 2a) [42].
Diverse indagini supportano il ruolo modulatorio dei prodotti ACE2, come Ang- (1–7), nel cervello. Non sorprende che l’espressione di Ang- (1-7) si trovi principalmente nei nuclei correlati alla regolazione della PA, come il tronco encefalico e l’ipotalamo, ed esercita effetti sinergici o antagonistici su Ang-II. È stato dimostrato che Ang- (1–7) modula la reattività del baroriflesso cardiaco [26].
È stato dimostrato che la sovraespressione dei suddetti membri centrali della RAS aumenta il livello della proteina ACE2 nell’OFS e influenza la sua espressione nel tronco cerebrale [43]. Pertanto, ACE2 sembra fornire un meccanismo compensatorio per limitare l’iperattività della RAS cerebrale.
In accordo con questo, la riduzione dell’mRNA di AT1R mediante un approccio di silenziamento genico è stata associata a una riduzione dell’mRNA di ACE2 nel tronco cerebrale [44]. Inoltre, ACE2 partecipa al metabolismo di alcuni peptidi non RAS, come i frammenti ipertesi di Apelin-13, neurotensina e bradichinina ipotensiva. Studi genetici sono stati mappati sul gene ACE2 in un locus di un tratto quantitativo correlato all’ipertensione sul cromosoma X. Inoltre, diversi studi hanno dimostrato una forte associazione del polimorfismo del gene ACE2 con ipertensione e cardiomiopatia ipertrofica, malattia coronarica e infarto del miocardio [45, 46].
Distribuzione di ACE2 nel cervello
Bassi livelli di mRNA di ACE2 sono stati dimostrati nel cervello umano utilizzando RT-PCR quantitativa in tempo reale due decenni fa [47]. Inoltre, la proteina ACE2 è stata osservata esclusivamente nelle cellule muscolari lisce endoteliali e arteriose dei vasi cerebrali mediante tecnica immunoistochimica [41].
Successivamente, studi in vitro utilizzando colture di cellule primarie cerebrali hanno riportato l’espressione di ACE2 prevalentemente nelle cellule gliali [48].
Al contrario, un altro studio ha mostrato la proteina ACE2 e l’mRNA principalmente nel citoplasma dei corpi cellulari neuronali dei topi. Inoltre, notevoli quantità di ACE2 sono state trovate anche nella sostanza gliale centrale e nel liquido cerebrospinale di campioni umani [49].
È stata dimostrata una distribuzione diffusa di ACE2 in tutto il cervello, specialmente nei centri coinvolti nella regolazione centrale della funzione cardiovascolare come i neuroni del tronco cerebrale, così come in aree non cardiovascolari come la corteccia motoria e il nucleo del rafe [50]. In un ulteriore lavoro, è stata confermata anche la presenza di ACE2 mRNA e proteine nel tronco cerebrale di topo [44].
Secondo i dati ottenuti dai topi transgenici, l’ACE2 era significativamente alto nell’OFS. Inoltre, è stata riportata una correlazione tra l’mRNA di ACE2 e l’espressione di varie proteine nel nucleo tractus solitario, nel nucleo vagale dorsale e nel midollo ventrolaterale caudale [50], suggerendo un ruolo cruciale di ACE2 nella modulazione del nervo autonomo sistema.
L’espressione di ACE2 è stata dimostrata anche nel sistema olfattivo e in altre importanti regioni cerebrali, come i ventricoli cerebrali e la substantia nigra, nonché aree che sono direttamente o indirettamente correlate alle vie olfattive, inclusi i nuclei ipotalamici, l’amigdala, l’ippocampo e la corteccia frontale [21].
ACE2 come recettore per SARS-CoV-2
Oltre al sistema olfattivo, ACE2 è notevolmente espresso dalle cellule epiteliali della mucosa del tratto orale, nasale e respiratorio [51, 52], a sostegno dell’idea che ACE2 potrebbe essere un potenziale recettore che media l’ingresso di SARS-CoV2 nelle cellule [2, 23, 53].
Come accennato in precedenza, l’ACE2 è stato rilevato nelle cellule gliali e nei neuroni di varie aree del cervello e attraverso l’ipotalamo e altri nuclei indirettamente potrebbe creare un collegamento aggiuntivo alla via olfattiva (Fig. 3a). Tuttavia, non è ancora noto come SARS-CoV-2 raggiunga esattamente il SNC attraverso la via olfattiva [23].
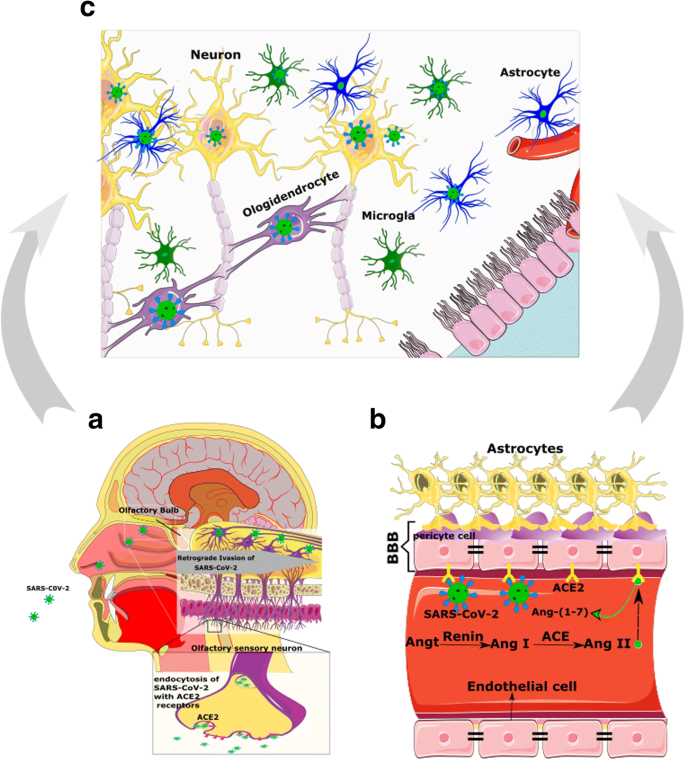
L’interazione tra la proteina spike virale e l’ACE2 è più complicata di una semplice interazione lock-and-key e altre presunte molecole sono coinvolte nel processo che consente a SARS-CoV-2 di invadere le cellule (Fig. 2b). In questa scena, Furin e la proteasi transmembrana serina 2 (TMPRSS2) sono due attori chiave [54].
La localizzazione di TMPRSS2 nella membrana cellulare innesca la proteina spike per facilitare l’assorbimento virale da parte di ACE2. Durante l’interazione virus / recettore, la proteina spike virale viene scissa da Furin [55, 56].
La co-espressione di ACE2 e TMPRSS2 nelle cellule epiteliali ma non nei neuroni sensoriali del sistema olfattivo suggerisce che l’anosmia indotta da SARS-CoV-2 e altri disturbi olfattivi siano correlati alle cellule non neuronali [57]. In contrasto con questi dati, uno studio utilizzando IHC e analisi geniche ha dimostrato che ACE2, TMPRSS2 e Furin sono co-espressi nella mucosa respiratoria e olfattiva, in particolare nelle cellule di supporto dell’epitelio olfattivo e nelle ghiandole di Bowman.
Questo studio ha anche rilevato ACE2 nei neuroni del recettore olfattivo insieme ai neuroni del bulbo olfattivo e ha concluso che il deterioramento del senso dell’olfatto può essere dovuto a una disfunzione neuronale.
Infine, hanno dichiarato che l’attacco diretto dei neuroni del recettore olfattivo da parte di SARS-CoV-2 è inaspettato, mentre queste cellule esprimono solo ACE2, ma non TMPRSS2 o Furin [54]. Pertanto, ad eccezione dell’infezione diretta dei neuroni olfattivi, dovrebbero essere prese in considerazione altre spiegazioni per anosmia o iposmia in pazienti con COVID-19 (Fig. 3) [21].
Allo stesso modo, sono stati rilevati ACE2 e TMPRSS2 a bassi livelli nelle cellule basali orizzontali umane (HBC) [57]. Gli HBC sono progenitori dell’epitelio olfattivo e vengono continuamente divisi per sostituire i neuroni sensoriali durante l’età adulta [58].
Quindi, i neuroni sensoriali olfattivi originati da HBC infetti possono essere caricati da SARS-CoV-2 e consegnare il virus alla corteccia olfattiva attraverso il bulbo olfattivo [21]. Va sottolineato che oltre al sistema olfattivo, non devono essere trascurate altre vie di ingresso nel SNC [23].
Considerando che ACE2 è espresso nell’endotelio vascolare, periciti, neuroni cerebrali, astrociti e oligodendrociti [23, 59], SARS-CoV-2 può rompere il BBB e invadere il sistema nervoso centrale attaccando il sistema vascolare. Inoltre, SARS-CoV-2 legandosi ad ACE2 può aumentare la pressione arteriosa nel cervello, aumentare la permeabilità di BBB e aumentare il rischio di ictus [60, 61] (Fig. 3B e Fig. 4).
Pertanto, oltre alla probabile invasione diretta delle cellule neurali da parte di COVID-19, l’esaurimento cronico dell’ACE2 cerebrale dovuto all’occupazione da parte di particelle virali e alla perturbazione dell’equilibrio di ACE / ACE2 può anche contribuire alle manifestazioni neurologiche di COVID-19.
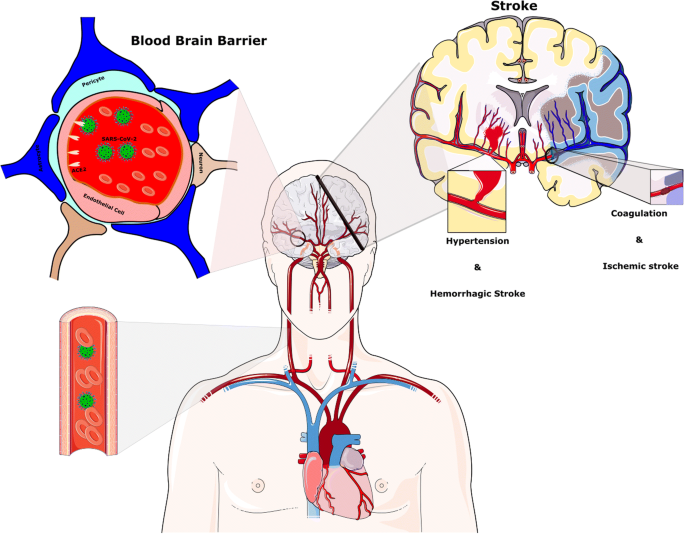
Ictus dopo l’infezione da SARS-CoV-2. È stato generalmente affermato che l’ipertensione è associata alla malattia SARS-CoV-2. La riduzione di ACE2 da parte di SARS-CoV-2 fornisce la condizione per l’ipertensione dipendente da Ang II. Livelli elevati di PCR e D-dimero indicano un alto stato infiammatorio e un’anomalia nella cascata della coagulazione e probabilmente giocano un ruolo chiave nella fisiopatologia dell’ictus nel contesto dell’infezione da COVID-19
ACE2 e Cytokine Storm, il meccanismo principale delle complicazioni COVID-19
In stato di salute, l’asse ACE2 / MasR inibisce le azioni pro-infiammatorie dell’asse ACE / Ang II / AT1R degradando Ang II insieme alla produzione di Ang- (1–7 ) e riduce l’espressione delle protein chinasi attivate dai mitogeni (MAPK), del fattore nucleare kappa B (NF-κB) e dei fattori infiammatori, come l’interleuchina-6 (IL-6), il fattore di necrosi tumorale α (TNFα) e IL -8 [62].
Al contrario, l’iperattivazione del RAS può indurre infiammazione dovuta al rilascio di citochine profibrotiche, come il fattore di crescita trasformante beta (TFG-β), attraverso il braccio AT1R / ACE. Inoltre, alti livelli di Ang II e iperattivazione di AT1R attivano la cascata del complemento, inclusi C5a e C5b-9 [63,64,65].
In condizioni patologiche, come l’infezione da COVID-19, dopo il legame della proteina spike virale all’ACE2 nei pneumociti di tipo II, il complesso virus / ACE2 sottoregola l’ACE2 intracellulare e di conseguenza aumenta l’infiammazione [62]. Come risultato della sottoregolazione dell’ACE2, i livelli sierici di Ang II e l’attività dell’asse Ang II / AT1R sono potenziati [62].
Inoltre, la sottoregolazione di ACE2 porta all’attivazione della via NF-κB attraverso la fosforilazione della sua subunità p65 e il potenziamento della produzione di IL-6, TNFa, IL-1B e IL-10. Anche la MAPK, che svolge un ruolo critico nel rilascio di varie citochine, tra cui IL-1, IL-10, IL-12 e TNFα, è modulata da Ang II [66].
Diversi studi clinici hanno indicato un marcato aumento delle citochine pro-infiammatorie, come IL-2, IL-6 e TNF-α, nei casi gravi e moderati di COVID-19 [67,68,69,70,71,72, 73].
Inoltre, lo stesso SARS-CoV-2 stimola il percorso NF-κB indipendente da ACE2 attraverso i recettori di riconoscimento del pattern. Com’era prevedibile, questa ondata di citochine nel siero innesca una tempesta di citochine che si traduce in sindrome da distress respiratorio acuto (ARDS) nei casi gravi di COVID-19 [62].
Inoltre, l’elevata produzione locale di citochine nel tessuto cerebrale può indurre infiammazione [73] e portare a caratteristiche neurologiche osservate nei pazienti COVID-19 [74]. Va anche menzionato che i componenti virali sono in grado di interagire con varie molecole, come i recettori di riconoscimento del pattern (p. Es., Recettori Toll-like), NF-KB, cascate del complemento, alcuni fattori di trascrizione e mediatori dell’apoptosi cellulare [62, 75] .
Dopo l’ingresso del virus nel sistema nervoso centrale, le vie di segnalazione varie all’interno o tra le cellule potrebbero essere modificate e i neurotrasmettitori cerebrali e gli ormoni locali potrebbero essere disregolati.
Riferendosi al fatto che l’ARDS indotta da COVID-19 è una conseguenza di una risposta infiammatoria incontrollata caratterizzata da tempesta di citochine, diversi esperimenti in vivo e in vitro indicano la probabile implicazione della P2X7 ionotropica purinergica in questo processo. Il P2X7R svolge un ruolo critico nella neuroinfiammazione attraverso i suoi potenti effetti stimolatori sull’inflammasoma NLRP3, che di conseguenza porta all’attivazione della caspasi-1, al rilascio di IL-1 β e IL-18 e alla down-modulazione dei macrofagi. Il SARS-CoV può anche attivare direttamente l’inflammasoma NLRP3. Il blocco farmacologico o l’abbattimento genetico del P2X7R ha ridotto sostanzialmente l’infiltrazione delle cellule infiammatorie, i livelli di citochine e il danno polmonare. Inoltre, in assenza di P2X7R, la morte dei macrofagi alveolari e il rilascio di pro-IL-1 β sono diminuiti in un modello in vivo di infiammazione polmonare. Quindi, il P2X7R è stato proposto come biomarcatore infiammatorio per l’infezione da COVID-19 [76], che può predire l’esito di complicanze neurologiche dei pazienti con infezione da COVID-19. Il recettore purinergico P2X7 è ampiamente distribuito nel cervello [77] ed è collegato ai cambiamenti infiammatori e neurodegenerativi.
Considerando l’audace azione dell’iperinfiammazione nella fisiopatologia della SARS-CoV-2, i farmaci antinfiammatori non steroidei e i corticosteroidi possono essere un trattamento efficace per alleviare i sintomi del COVID-19. Prove emergenti hanno dimostrato l’efficacia dei corticosteroidi nel controllo del danno acuto indotto da COVID-19.
Tuttavia, la possibilità di rebound virale o altri effetti collaterali nella somministrazione a lungo termine di corticosteroidi non poteva essere esclusa [78]. A questo proposito, uno studio controllato randomizzato ha riportato una maggiore concentrazione di SARS-CoV-2 RNA nella settimana 2/3 di infezione in quelli trattati con corticosteroidi.
Inoltre, in un modello suino di infezione da CoVs respiratoria, alcune dosi di desametasone nella fase acuta dell’infezione hanno diminuito la risposta pro-infiammatoria acuta, ma con una somministrazione prolungata vi era il rischio di replicazione virale. Tuttavia, alcune prove supportano gli effetti benefici della limitata applicazione di farmaci antinfiammatori nella fase iniziale dell’infezione da COVID-19 [78].
Prove crescenti implicano la comorbilità dell’ipertensione e di altre forme di malattie cardiovascolari nei casi gravi di COVID-19 che sono comunemente trattati con ACEI e ARB. Teoricamente, gli ACEI e gli ARB dovrebbero aumentare il livello di ACE2 e successivamente l’ingresso di SARS-CoV-2 nelle cellule e portare a maggiori esiti avversi, come il danno polmonare. Tuttavia, secondo gli studi sperimentali, ACE2 era protettivo contro i danni ai polmoni. ACE2 forma Ang- (1–7) anti-infiammatorio da Ang II e quindi antagonizza l’azione infiammatoria di Ang II.
Inoltre, gli ACEI attraverso la riduzione della formazione di Ang II e ARB attraverso l’inibizione dell’attività di Ang II e il blocco di AT1R potrebbero controllare la risposta infiammatoria sistemica. Con i meccanismi sopra menzionati, questi agenti potrebbero prevenire lo sviluppo di ARDS, miocardite o danno renale acuto, che si verificano comunemente nel COVID-19.
Inoltre, gli ARB sono potenziali candidati per il trattamento delle complicanze della SARS-CoV-2. Tenendo conto che un livello elevato di ACE2 solubile nel siero può bloccare la proteina spike virale e ridurre l’assorbimento di SARS-CoV-2 da parte degli organi che esprimono ACE2, l’applicazione dell’ACE2 ricombinante è proposta come approccio terapeutico per COVID-19 [79].
Oltre agli ACEI e ai bloccanti dell’AT1R [79], i farmaci antinfiammatori, come il tiazolidinedione e l’ibuprofene, possono aumentare la quantità di ACE2 [80]. Non è chiaro se la prognosi infausta dell’infezione da COVID-19 nei pazienti ipertesi e diabetici sia correlata alle loro precedenti farmacoterapie e aggiustamenti molecolari o al loro profilo ACE2.
Secondo Fang et al., La vulnerabilità di un individuo al COVID-19 potrebbe essere il risultato sia della precedente terapia che dei polimorfismi del gene ACE2 [80]. Nel loro insieme, né il ruolo esatto dell’ipertensione né gli effetti benefici o dannosi degli ACEI o degli ARB [79] insieme agli agenti antinfiammatori sugli esiti di COVID-19 sono stati chiariti e devono essere ulteriormente esplorati.
Altri possibili meccanismi
Esistono diverse possibilità per quanto riguarda i meccanismi fisiopatologici mediante i quali COVID-19 influisce sulla funzione cerebrale. Un potenziale meccanismo per i meccanismi neurologici potrebbe essere correlato all’ipossia indotta da SARS-CoV-2. L’invasione virale disturba lo scambio di gas dal sistema respiratorio e porta all’ipossia generale. L’ipossia induce il metabolismo anaerobico nei mitocondri delle cellule cerebrali con conseguente sovrapproduzione di acido.
Livelli elevati di acido causano vasodilatazione intracerebrale, edema cerebrale, ostruzione del flusso sanguigno cerebrale, ischemia e mal di testa. Come risultato dell’ipossia continua, può comparire ipertensione intracranica. In pazienti ad alto rischio con malattie cardiovascolari, l’ipossia può anche indurre il verificarsi di ictus ischemico acuto e sintomi neurologici (Fig. 4) [2].
In definitiva, va detto che le complicazioni neurologiche potrebbero essere dovute a un’infezione secondaria, non al coronavirus stesso. Sebbene il BBB sia danneggiato a causa dell’infezione virale, è più facile per altri patogeni raggiungere il SNC. Pertanto, le infezioni intracraniche secondarie possono causare il coinvolgimento neurologico nei pazienti infetti da COVID-19 [2].
Manifestazioni neurologiche dell’infezione da COVID-19
Dopo aver riassunto i cambiamenti fisiopatologici rilevanti dell’infezione da COVID-19 che possono portare a manifestazioni neurologiche, descriviamo le complicanze neurologiche più comuni osservate negli individui infetti.
Iposmia
Una delle prime caratteristiche comuni della contaminazione da COVID-19 è l’iposmia [81]. È prevedibile la connessione diretta dei bulbi olfattivi con il cervello, la penetrazione di COVID-19 nel cervello. Sebbene un possibile meccanismo sia una trasmissione retrograda di COVID-19, attraverso l’epitelio olfattivo al cervello [82], non dovrebbero essere trascurati altri probabili percorsi.
Si ricorda che i precursori neuronali originati dalla zona subventricolare (SVZ) migrano e maturano attraverso il sistema migratorio rostrale fino a raggiungere il bulbo olfattivo. Il BBB di SVZ è più penetrabile rispetto ad altre aree del cervello a causa della mancanza di processi periciti e delle estremità degli astrociti, che avvolgono i vasi sanguigni [83].
D’altro canto, SARS-CoV-2 si lega a ACE2 e quindi potrebbe degradare Ang II in Ang- (1–7) e aumentare il livello di Ang II nel flusso sanguigno. È stato riferito che un alto livello di angiotensina può evocare effetti apoptotici sulle cellule staminali neurali [84] e ridurre il numero di precursori nella SVZ e successivamente diminuire il numero di cellule che migrano verso il bulbo olfattivo. Come risultato di una riduzione della sostituzione di nuovi neuroni nel bulbo olfattivo, l’olfatto può essere disturbato.
Convulsioni
Un’altra caratteristica neurologica osservata nei pazienti con COVID-19 sono le convulsioni [19]. In uno studio condotto su 70 soggetti con infezione da MERS-CoV, il sequestro è stato segnalato nel 9% dei pazienti [85]. Inoltre, si sono verificate convulsioni in alcuni pazienti con COVID-19 [11]. La RAS ha un ruolo vitale in diverse condizioni neurologiche, comprese le convulsioni. L’inibizione costante del sistema renina-angiotensina ha prevenuto le convulsioni in un modello di epilessia di ratto [86].
Inoltre, l’espressione dei recettori Ang II, AT1R e AT2R nella formazione ippocampale dei pazienti con epilessia del lobo temporale è sovraregolata, supportando il potenziale coinvolgimento della RAS nell’innesco delle crisi [87]. La riduzione della disponibilità di ACE2, così come il miglioramento dei valori di Ang II e dei suoi mediatori pro-infiammatori a valle in COVID-19, possono contribuire al verificarsi di convulsioni.
Le crisi epilettiche possono aumentare la produzione di citochine, come IL-1b e TNFα, che a loro volta regolano la patogenesi e il decorso degli attacchi convulsivi [10].
Ictus
Fino ad ora, l’ictus è uno degli esiti avversi più gravi e fatali dell’infezione da COVID-19. Diverse indagini hanno riportato il verificarsi di malattie cerebrovascolari acute, in particolare ictus ischemico in pazienti con grave infezione da COVID-19 [88,89,90].
Queste gravi complicanze di COVID-19 sono dovute a eventi trombolitici, difetti della parete vascolare ed emorragia cerebrale. Circa il 6% dei pazienti con COVID-19 grave ha sviluppato l’insulto cerebrovascolare nel corso della malattia [11]. Avula et al. hanno riferito che quattro pazienti con infezione da SARS-CoV-2 confermata mediante PCR hanno mostrato una verifica radiografica di ictus acuto [91].
Inoltre, un soggetto ricoverato in ospedale per un ictus sottile nascosto con debolezza dell’arto destro con scarsa fluidità del linguaggio è stato diagnosticato con COVID-19 [30].
L’aumento dei livelli di CRP e D-dimero, che indica un alto stato infiammatorio e un’anomalia nella cascata della coagulazione, probabilmente svolgono un compito funzionale nella fisiopatologia dell’ictus nel contesto dell’infezione da COVID-19 [53]. L’iperattivazione della cascata del complemento nel processo della risposta infiammatoria da parte di SARS-CoV-2 può portare a trombosi (Fig. 4) [92, 93].
Inoltre, a causa della riduzione delle proteine ACE2 e di un ulteriore aumento dell’Ang II, un aumento della pressione arteriosa può aumentare il verificarsi di ictus. In accordo con questa affermazione, l’occlusione dei grandi vasi, come l’arteria cerebrale media, era correlata con l’infezione da CoVs [94]. Inoltre, un aumento dell’espressione di CD147 negli astrociti è stato riportato in un modello sperimentale di ictus [16].
Atassia
L’infarto cerebellare acuto può causare letargia, disartria e atassia [95]. Inoltre, l’atassia cerebrale acuta può coincidere con condizioni infiammatorie immuno-mediate ed encefalopatie [96], che sono comuni nell’infezione SARS-CoV-2.
Cefalea La
cefalea è un segno comune di vari problemi medici, come l’infezione virale e l’ipertensione arteriosa. La cefalea indotta dall’ipertensione è solitamente bilaterale e acuta ed è collegata a un forte aumento della pressione arteriosa diastolica ≥ 120 mmHg o della pressione sistolica ≥ 180 mmHg [97,98,99].
L’equilibrio tra ACE2 e ACE è importante per la regolazione della BP [100]. L’alterazione del rapporto ACE / ACE2 in COVID-19, quindi, può aumentare la pressione arteriosa e innescare mal di testa. Un’altra causa plausibile di mal di testa nei malati di COVID-19 potrebbe essere l’ondata di citochine infiammatorie, tra cui TNFα, IL-1 e IL-6 [101, 102]. L’ipossia intracerebrale e l’infezione secondaria del tessuto cerebrale sono altre potenziali cause di mal di testa [2].
Encefalopatia
Come la maggior parte delle condizioni descritte, l’encefalopatia non è una malattia specifica, ma un sintomo o una sindrome accompagnata da una disfunzione cerebrale generale causata da radici organiche o non organiche. A causa del coinvolgimento di vari organi nell’infezione da SARS-CoV-2, il rischio di encefalopatia è molto alto [103]. Esistono diversi tipi di encefalopatia, come encefalopatia ipertensiva, encefalopatia ipossico-ischemica, encefalopatia epatica ed encefalopatia uremica che possono essere osservati in pazienti con COVID-19.
Encefalopatia
ipertensiva La crisi ipertensiva avrebbe potuto provocare mal di testa, convulsioni e disturbi della coscienza [104]. L’encefalopatia ipertensiva si verifica a causa della rottura dell’integrità del BBB a seguito di un’elevata pressione arteriosa che porta a iper-perfusione cerebrale e, infine, edema cerebrale.
In uno stato normale, il meccanismo autoregolatorio cerebrale protegge il BBB da pericolosi aumenti di pressione sistolica [105]. Al contrario, accelerazioni notevolmente rapide della pressione arteriosa provocano perdite di plasma ortostatico sui capillari [106]. È stato generalmente affermato che l’ipertensione è associata alla malattia SARS-CoV-2 [107].
La riduzione di ACE2 da parte di SARS-CoV-2 aumenta il rischio di ipertensione dipendente da Ang II [108]. Ciò può innescare una cascata che provoca l’encefalopatia ipertensiva attraverso l’interruzione dell’integrità del BBB e l’iper-perfusione cerebrale.
Encefalopatia
ipossico-ischemica La lesione cerebrale ipossico-ischemica o encefalopatia (HIE) è una sindrome neurovascolare e neuro-metabolica. HIE nasce come conseguenza di una mancanza di apporto di ossigeno o glucosio o di un metabolismo cerebrale modificato dopo un grave danno cerebrale.
Questa encefalopatia è il risultato di una ipo-perfusione o iperossigenazione globale piuttosto che di un infarto vascolare specifico in un’area cerebrale [109]. Alcune regioni cerebrali tra l’arteria cerebrale anteriore e quella media possono essere influenzate dall’HIE, inclusi i gangli della base, la formazione dell’ippocampo, il cervelletto e il talamo [110].
La disfunzione respiratoria a seguito di infezione virale potrebbe anche evocare ipossia generale e quindi indurre il metabolismo anaerobico nei mitocondri delle cellule cerebrali. Vasodilatazione intracerebrale a basso pH, edema cerebrale, ostruzione del flusso sanguigno cerebrale e ischemia.
In pazienti ad alto rischio con malattie cardiovascolari, l’ipossia può anche terminare con ictus ischemico acuto e altri sintomi neurologici [2, 93]. Come accennato, un’eziologia comune di HIE sono le lesioni cardiovascolari, che sono reciproche tra i pazienti con infezione da SARA-CoV-2 [111]. L’ACE2 è anche ampiamente espresso nelle cellule endoteliali coronariche, nei fibroblasti cardiaci e nei cardiomiociti [112]. La sovraespressione di ACE2 esercita una funzione protettiva e inverte l’insufficienza cardiaca, mentre la soppressione dell’espressione di ACE2 può accelerare il progresso dello scompenso cardiaco [113].
Encefalopatia
epatica Sono state segnalate anomalie epatiche e persino insufficienza epatica dopo infezione da COVID-19. I danni epatici potrebbero essere dovuti all’effetto diretto di SARS-CoV-2 sul fegato o agli effetti collaterali dei farmaci [114]. Xu e colleghi hanno riportato steatosi e danno epatico in un paziente con SARS-CoV2 [23]. La disfunzione cardiovascolare potrebbe essere un’altra potenziale causa di encefalopatia epatica correlata a COVID-19.
Encefalopatia
uremica L’encefalopatia uremica potrebbe essere una conseguenza dell’insufficienza renale acuta o cronica dopo una riduzione della velocità di filtrazione glomerulare, che è frequentemente segnalata nei pazienti con COVID-19. Ciò provoca una sindrome con vari sintomi, come affaticamento o convulsioni [115, 116].
Alcuni studi hanno riportato un’incidenza amplificata di danno renale acuto a seguito di SARS-CoV-2. Non è noto che questo fallimento sia causato da reazioni infiammatorie evocate da virus o disturbi cardiovascolari [117,118,119]. Il rene ha un ruolo essenziale nel controllo dell’equilibrio acido-base ed è anche un regolatore cruciale dell’omeostasi cerebrale attraverso l’escrezione di tossine e la modifica delle concentrazioni di citochine. L’encefalopatia uremica si verifica apparentemente a seguito di uno squilibrio ACE / ACE2 durante l’infezione da COVID-19 [120, 121].
Conclusione
RAS e ACE2 svolgono un ruolo cruciale nella patogenesi delle manifestazioni neurologiche dell’infezione da COVID-19, presumibilmente attraverso i loro effetti regolatori sulla circolazione sanguigna e sulla PA. Inoltre, l’ACE può implicare sintomi neurologici correlati a SARA-CoV-2 attraverso i suoi effetti modulatori nelle risposte infiammatorie e immunitarie [119].
Secondo il crescente corpo di prove, gravi complicanze neurologiche dell’infezione da COVID-19, come l’ictus, peggiorano gli esiti di COVID-19. Sono necessari ulteriori studi per indagare se la manipolazione farmacologica di RAS e ACE2 possa influenzare il decorso e gli esiti di COVID-19.
link di riferimento: https: //link.springer.com/article/10.1007/s12035-020-02149-0



{kind=link}