









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Melbourne researchers are working towards a potential treatment to slow the progression of motor neuron disease (MND), offering hope to people with this debilitating and incurable illness.
The research team have uncovered how inflammation in MND is triggered. Pinpointing the molecules involved in this pathway could be a first step towards a new treatment for MND.
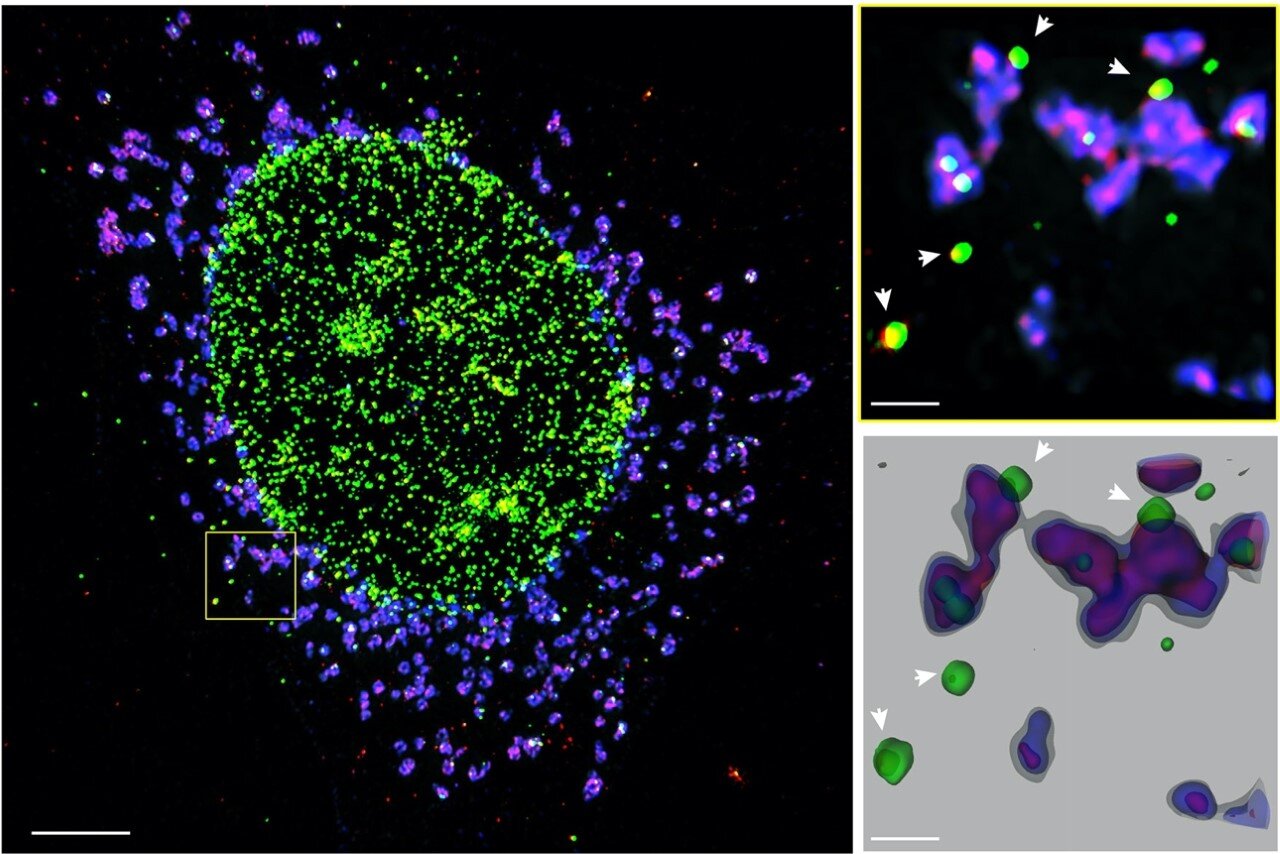
They found that by blocking an immune sensor called STING, they could dramatically prevent inflammation from MND patient cells, paving the way for a new class of drugs to be developed for people with neurodegenerative disorders, such as MND.
The discovery, published today in Cell, was led by Walter and Eliza Hall Institute researchers Associate Professor Seth Masters and Dr. Alan Yu, with colleagues from the University of Melbourne and Hudson Institute.
Halting the inflammatory response
MND is an incurable condition in which the nerve cells controlling the muscles that enable us to move, speak, swallow and breathe, fail to work. One in 10,000 Australians will be diagnosed with MND in their lifetime and the average life expectancy from diagnosis is just two years.
Most people suffering from MND have an accumulation of a protein called TDP-43 within cells of the central nervous system. This build-up is associated with an inflammatory response that precedes major symptoms of MND.
Institute researchers investigated how the disease-causing inflammation is triggered in MND, said Associate Professor Masters. “This unexpectedly identified that an immune sensor called STING is activated downstream of TDP-43. Fortuitously, our team had already studied the role of STING in other inflammatory diseases and are now working out how to block it.”
The team then used new inhibitors – drug-like compounds – to block different components of this inflammatory pathway.
“Using cells from patients with MND that we can turn into motor neurons in a dish, we showed that blocking STING dramatically prevented inflammation and kept the cells alive longer. This is an exciting first step before taking these inhibitors into the clinic for treatment for MND.
Vital first step towards a treatment
Associate Professor Masters said his research had also established activation of STING in people who had passed away due to MND.
“We are now aiming to validate a biomarker of the pathway earlier in the disease progression. Once this neuroinflammatory biomarker is validated, we will better understand which patients will benefit the most from treatments targeting the pathway,” he said.
“With this knowledge, there is the potential to develop a treatment for patients with MND.
“Interestingly, our preclinical models suggest that although the anti-inflammatory drugs that inhibit STING did not prevent disease onset, they did slow the degenerative progression of disease.”
Hope for people with MND and other neurodegenerative disorders
Associate Professor Masters said this discovery offered hope for people diagnosed with the debilitating condition.
“We are hopeful this research could lead to a treatment for people with established MND, who currently have very few treatment options and a life expectancy post diagnosis of just two to five years,” he said.
“While it isn’t a cure, we hope it might extend life expectancy and dramatically improve the quality of life for people diagnosed with MND.”
Associate Professor Masters said a future treatment might also be effective in slowing the progression of other neurodegenerative disorders.
“We are hoping to develop a new class of drugs that would act as STING inhibitors to stop the progression of neurodegenerative disorders, such as MND, Frontotemporal Dementia and Parkinson’s disease.”
Motor neuron disease (MND) is said to be a progressive neurological disorder that presents with both lower motor neurons (anterior horn cells project from the brainstem and the spinal cord to the muscle) and upper motor neuron signs (neurons that project to the brainstem and spinal cord from higher cortical centers).[1]
While the anterior horn cell and the corticospinal tract have been shown to be the primary site of involvement, the involvement of other parts of the nervous system (cortical, autonomic, cerebellar and extrapyramidal system) has also been documented.[1] The four main phenotypes of motor neuron disease, based upon the site of origin and the severity of neurological involvement, are as follows: Amyotrophic lateral sclerosis, progressive bulbar palsy, progressive muscular atrophy, and primary lateral sclerosis.[2]
Other systems used for the classification of this disease have used the site of onset (spinal or bulbar onset), degree of adherence to the El-Escorial (and Airlie house) criteria and pattern of heritability (sporadic versus familial) as criteria for characterizing this neurodegenerative illness with a complex genetic basis.[3] MND has been shown to be a disease of middle age with a mean age of 58 to 63 years at the time of onset for Sporadic Amyotrophic lateral sclerosis (ALS), and 40-60 years of age for familial ALS.[4] There has been a growing ethical debate along with pleas to formulate policies pertaining to the provision of Euthanasia and physician-assisted suicide, which have brought this disease into public focus.[5]
Etiology
Existing research points towards an underlying genetic basis. Four genes are associated with up to 70 percent cases of Familial ALS, namely the C9ORF72, TARDBP, SOD1, and FUS. Over 25 genes and loci have been identified in relation to the predisposition of the disease, amongst which the C9ORF72 (Chromosome 9 open reading frame), has been associated with 40 percent cases of familial and 10 percent cases of sporadic MND.
This mutation, which leads to a hexanucleotide repeat expansion, has also been found in cases presenting which Fronto-temporal dementia which has lead to a hypothesis that these neurological disorders exist as part of a dynamic spectrum of neurological syndromes.[6] This observation also provides evidence for the involvement of extra motor structures in MND spectrum disorders.
While this particular locus has less than 30 repeats in healthy individuals, patients with C9 related ALS, have a c9orf72 expansion mutation that has hundreds to thousands of repeats.[7] The presence of this expansion leads to ALS in two different ways. For example, TDP-43 accumulation and repeat-associated non-ATG translation of nuclear RNA foci leading to the formation of repeat derived RNA and dipeptide repeat proteins, which have been postulated to cause neurotoxicity.[8]
TARDBP gene encodes the TAR DNA-binding protein 43, SOD-1 gene encodes for superoxide dismutase. In contrast, the FUS gene codes for the similarly named (Fused in sarcoma) RNA binding protein.[8]
A study of population-based registries of ALS has led a degree of credibility to the hypothesis that ALS follows a complex genetic inheritance, where a variety of environmental factors interact with genetic mutations (present in “at-risk” variants), through a multistep process that decides the pattern of the disease manifestation. The occurrence of rare variants makes extensive genome-wide studies less suited for the study of this disease.[9]
Dysregulation of micro RNA and variations in ion channels that predispose to cellular excitotoxicity has also been postulated as the etiology underlying motor neuron disease.[10]
Familial forms of ALS are also characterized by penetrance of less than 50 percent and genetic pleiotropy (where a single gene can lead to the manifestation of multiple phenotypical traits). In contrast, sporadic forms have been associated with oligogenic (determination of a phenotypic trait by more than one gene) and polygenic inheritance.[11]
Among environmental and lifestyle factors that have been associated with the development of ALS, cyanotoxins, and related compounds deserve mention. The association between the ALS and pre-morbid physical prowess is also an area where some research has been done.
An increased incidence in athletes, who have been exposed to repetitive head trauma was proposed, and discarded subsequently, due to the absence of evidence.[12] Amongst other environmental factors, cigarette smoking, and past history of military service have been consistently shown to be associated with an increased propensity for developing ALS.[4]
Treatment / Management
Disease-modifying Therapies
Riluzole, an NMDA (N-methyl-D-aspartate) receptor antagonist, reduces glutaminergic transmission by acting on the presynaptic voltage-gated sodium channels. Relative glutamate excess has been shown to lead to upper and lower motor neuron excitotoxicity contributing to neuronal cell death.[54] It has been shown to be associated with a statistically significant improvement in tracheostomy free survival.[55]
Though sustained use for 18 months was not associated with improved muscle strength, it has been shown to improve survival by three months (equitable to a 9 percent increase in one-year survival).[56] It was found to be very useful in those with moderate functional impairment in those with advanced disease when used on a compassionate basis.[57]
The standard dose of Riluzole in the management of ALS is 50 mg two times daily.[58] The most common adverse effects include nausea, asthenia, and derangement in liver enzymes.[57] Pancreatitis and fulminant hepatic failure have also been reported as rare adverse effects.[59] Regular blood testing every month for the first three months, followed by three monthly for nine months and yearly, is recommended.[60] An increase of over three times the upper limit of normal in hepatic enzymes can be managed by dose reduction, followed by withdrawal followed by the re-introduction of the drug[60]. Riluzole is contraindicated in hepatic and renal impairments and in those who are breastfeeding and lactating. Further trials have been advised to test the efficacy of different dosing regimens.[61]
Edaravone has also been shown to impede disease progression in those patients with early-onset disease and rapid disease progression.[62] Its role in the management of all patients with ALS remains to be addressed.[63] The drug acts as an antioxidant, which has been shown to prevent the nitration of tyrosine residues in experimental animal models.[64]
Symptom Directed Treatment
Spasticity:
Spasticity may be present in most of the patients with primary lateral sclerosis, though it may only be present with a lesser frequency in patients with amyotrophic lateral sclerosis.[65] Tools to measure spasticity have included the modified Ashworth scale, numerical rating scale spasticity responses, and the Rasch based scale for patient-reported spasticity, among others.[66] Gabapentin, baclofen, tizanidine, benzodiazepines, and levetiracetam have been advised for the management of spasticity.
The practice of these drugs is limited by the occurrence of fatigue and weakness.[67] Baclofen may sometime induce hyperpolarisation of some of the afferent terminals and also inhibits monosynaptic and polysynaptic reflexes usually at the spinal level.[68] Intrathecal baclofen, wherein the drug is infused into the cerebrospinal fluid through a surgically placed infusion pump, has also been used in the management of spasticity in primary progressive aphasia.[69] Stages of the disease.[16]
Siallorhoea is caused by the inability to swallow saliva, which may be the result of tongue spasticity, weakness of the facial, mouth and pharyngeal muscles, loss of pharyngeal coordination, and function.[70] Anticholinergic drugs such as atropine, hyoscine, glycopyrrolate, and amitryptiline have been found to be beneficial in the management of sialorrhea.
Salivary gland irradiation and botulinum toxin injections have also been proposed as treatment modalities.[16] Anticholinergic drugs have been associated with urinary retention and constipation.
They are contraindicated in patients with heart block and benign prostatic hypertrophy.[1] Optimum advice on diet, swallowing, positioning, posture, hydration, and oral care should be provided. Nebulizers, humidifiers, and mucolytics should be considered.[71]
Pain:
15% to 85% of patients, more commonly nociceptive than neuropathic in nature. Later stages of the ailment may be characterized by musculoskeletal pain, which arises as a result of the loss of protective sheath comprising of muscles that protect the bones and joints. Muscle contractures and bone stiffness may also be a source of pain. Decubitus ulcers seen in 16 percent of patients may also be associated with pain.
Increased intensity of pain in the later stages of the disease has also been associated with assisted suicide.[72] While, NSAIDs, opioids, and cannabinoids have been used for nociceptive pain, gabapentinoids, and tricyclic antidepressants have been used in neuropathic pain. The dearth of randomized data on this topic is a matter of some concern. Intra-articular injections of lidocaine, along with physical therapy and assistive range of motion exercises, have also been helpful.[72]
Muscle cramps:
Cramps are caused by the instability of motor units.[1] Quinine sulfate, mexiletine, and levetiracetam have been used in ameliorating these symptoms. Caution is advised with the use of quinine sulfate as it has been associated with QT prolongation, arrhythmias, and bradycardia.
Dysphagia:
Strategies to mitigate this particularly distressing symptom include dietary changes. That consists of the change in consistency of the diet, high calories, and high protein diets, fluid thickeners along with exercises to facilitate swallowing. Insertion of a percutaneous gastrostomy tube for enteral nutrition has also been advised in those with a weight loss in excess of five percent and in whom swallowing can be associated with the occurrence of dangerous complications. In those in whom these procedures are contraindicated or those with respiratory insufficiency, feeding through a central venous catheter should be tried.[1]
Dysarthria:
Up to 30 percent of the patients with ALS develop dysarthria. This might extend to 80 percent of patients during the course of the disease.[1] Speech therapy, use of customized software for augmentative and alternative software have been shown to enhance the quality of life in ALS.[73]
Coagulation abnormalities (Deep venous thrombosis):
The annual incidence of coagulation abnormalities has been in the range of 3 and 11 percent. The use of prophylaxis, compression stockings, and standard doses of anticoagulation as per existing guidelines have been advised.[74]
Neuropsychiatric disturbances:
Neuropsychiatric disturbances such as pseudobulbar affect and depression have been reported.[75] The pseudobulbar effect has been postulated to be caused due to the disruption of the corticopontocerebellar pathways. Impaired volitional control and cerebellar demodulation have been considered central to the neuropathogenesis of this symptom.[76] It has also been described as dysmetria caused due to cerebellar dysmodulation.
Depression may be an early manifestation of frontal lobe dysfunction and may signify widespread cortical involvement.[77] Tricyclic depressants and selective serotonin reuptake inhibitors (SSRIs) have been used in the management of depression. Pseudobulbar effects have been managed with the use of these antidepressants.[78] Dextromethorphan and quinidine have also been shown to be useful in the management of this symptom.
Though no specific agents have been found to be useful in the treatment of cognitive symptoms, SSRIs have been found to be helpful in controlling symptoms such as control of loss of inhibition, overeating, and compulsive behaviors. In contrast, antipsychotics have been found helpful in the treatment of restlessness.
Respiratory symptoms:
The weakness of respiratory muscles may present in the form of the loss of ability to cough and respiratory insufficiency.[79] The monitoring of physiological parameters of respiratory function has been advised as respiratory symptoms have been found to poorly correlate with respiratory muscle function.[80]
Supine Forced vital capacity (FVC) may be used as a marker of diaphragmatic weakness.[81] Methods to assist the patient while coughing (cough assist devices such as physiotherapy maneuvers like tussive squeeze, mechanical insufflator – exsufflator devices) have been advised.[82]
The usage of non-invasive ventilation (NIV) has been associated with improvement in parameters of respiratory function, which is also associated with an improvement in the quality of life, along with a substantial increase in survival.
The presence of dyspnoea, orthopnoea, and daytime fatigue, vital capacity less than 80 percent of the normal, partial pressure of CO2 more than 45 mm of Hg, nocturnal hypoventilation (when the oxygen saturation is less than 88-90 percent for more than 5 percent of the time is asleep), maximal inspiratory pressure less than 60 percent predicted, sitting fixed vital capacity less than 50 percent, sniff nasal inspiratory pressure less than 40 percent have been listed as various indications for non-invasive ventilation.
NIV is poorly tolerated in 30 percent of patients due to anxiety, emotional lability from pseudobulbar damage, hypersalivation, claustrophobia, and soreness of the nasal bridge.[83] The presence of bulbar dysfunction at the onset and cognitive impairment has been shown to correlate with poor compliance.[84]
Persistent respiratory insufficiency, despite the use of NIV and severe bulbar dysfunction, has been reported as indications for tracheostomy.[85]
The use of diaphragmatic muscle pacing, where four electrodes are placed on the motor roots of the phrenic nerve on the abdominal surface of the diaphragm has also been studied.[86] The procedure involves the implantation of intramuscular electrodes on the abdominal surface of the hemidiaphragm at motor points.
Intramuscular diaphragmatic pacing aims to make the diaphragm contract, strengthen the force of contraction, and lead to a decrease in the patient’s dependence upon mechanical ventilation. After implantation, the patient undergoes a conditioning program that involves gradual weaning from the ventilator.
Current evidence suggests long term safety concerns related to the occurrence of adverse events such as capnothorax or pneumothorax, acute respiratory failure requiring mechanical ventilation, venous thromboembolism, and gastrostomy tube placement. Suture granuloma, infection at the stimulation cable entry point, and superficial wound infection has also been reported.[86]
Fatigue:
Fatigue has been reported as a frequent and debilitating symptom characterized by reversible weakness. It is characterized by a feeling of tiredness, which is not relieved by rest. Both pharmacological measures (Modafinil) and non-pharmacological measures (resistance exercise, respiratory exercise, and repetitive transcranial magnetic stimulation) have been used in the management of fatigue. The quality of evidence for the effectiveness of these interventions has been reported to be very low, and more research is needed on this topic.[87]
Palliative Medicine Interventions
Palliative medicine interventions include discussions surrounding advanced care planning, provision of quality home-based care, adequate symptom control, prognostication about goals of care, liaison with organizations providing community palliative services, degree of aggressiveness of treatment, anticipatory prescribing, provision of support to family members (addressing caregiver burden), and bereavement support.
Formal care at the end of life should incorporate measures to improve the quality of life (such as a system to enable effective communication) and focus upon the management of commonly encountered symptoms.The use of opioids and benzodiazepines for breathlessness, glycopyrrolate for increased secretions, antipsychotics for delirium, and other non-pharmacological treatment options may be indicated in patients approaching their end of life.[88]
Reduction in ICU utilization at the end of life, number of deaths in the hospital setting, and costs of treatment are essential indicators of provision of high-quality care.[89] For patients being treated in the intensive care setting, discussions centered upon withdrawal of life-sustaining measures and DNCPR (Do not perform cardiopulmonary resuscitation) decisions may be indicated.[90]
Physiotherapy
Exercise programs such as structured exercise programs may improve stiffness, prevent contractures, reduce discomfort, and optimize the quality of life. Provision of the orthosis, if necessary, should be facilitated without delay.[91]
Communication
Communication should be assessed by a speech and language therapist. Constant review during interdisciplinary meets is advised. The provision of augmentative and alternative communication strategies that cause the least interference with the patient’s quality of life is recommended.[92]
Future therapeutic options
Use of antisense oligonucleotides in SOD 1 related and C9orf related ALS are subjects of research in future clinical trials.[93]
Use of stem cell therapy
The proposed mechanism of the utility of stem cell therapy includes the differentiation of stem cells into spinal neurons that synapse with existing motor neurons to establish or maintain neurocircuitry and provide neurotrophic support. Differentiation of stem cells into non-neuronal cells can also impact disease progression by providing neurotrophic support, preventing oligodendrocyte dysfunction and toxicity. Peripheral stem cell transplantation can maintain the neuromuscular junction.
The immunomodulatory effect of mesenchymal stem cells mobilized from the bone marrow can also attenuate the inflammatory response via the production of anti-inflammatory mediators.
The commonly used stem cell therapies include olfactory ensheathing stem cells, endogenous mesenchymal stem cells, autologous mesenchymal stem cells, and neural progenitor cells. The most significant number of trials of stem cell therapy involves the use of mesenchymal stem cells.[94]
rewference link : https://www.ncbi.nlm.nih.gov/books/NBK560774/
More information: Chien-Hsiung Yu et al, TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS, Cell (2020). DOI: 10.1016/j.cell.2020.09.020



{kind=link}