










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



People with multiple sclerosis (MS) gradually develop increasing functional impairment. Researchers at Karolinska Institutet have now found a possible explanation for the progressive course of the disease in mice and how it can be reversed.
The study, which is published in Science Immunology, can prove valuable to future treatments.
MS is a chronic inflammatory disease of the central nervous system (CNS) and one of the main causes of neurological functional impairment.
The disease is generally diagnosed between 20 and 30 years of age.
It can cause severe neurological symptoms, such as loss of sensation and trembling, difficulties walking and maintaining balance, memory failure and visual impairment.
MS is a life-long disease with symptoms that most often gradually worsen over time.
In the majority of cases the disease comes in bouts with a certain amount of subsequent recovery. A gradual loss of function with time is, however, inevitable.
Research has made great progress in treatments that reduce the frequency and damaging effects of these bouts.
“Despite these important breakthroughs, the disease generally worsens when the patient has had it for 10 to 20 years,” says Maja Jagodic, docent of experimental medicine at the Department of Clinical Neuroscience and the Centre for Molecular Medicine, Karolinska Institutet.
“There is currently only one, recently approved, treatment for what is called the secondary progressive phase. The mechanisms behind this progressive phase require more research.”
Researchers at Karolinska Institutet have now shown that recovery from MS-like symptoms in mice depends on the ability of the CNS’s own immune cells – microglia – to break down the remains of damaged cells, such as myelin.
The processes was interrupted when the researchers removed a so-called autophagy gene, Atg7. Autophagy is a process where cells normally break down and recycle their own proteins and other structural components.
Without Atg7 the ability of the microglia to clean away tissue residues created by the inflammation was reduced. These residues accumulated over time, which is a possible explanation for the progressiveness of the disease.
The study also shows how microglia from aged mice resemble the cells from young mice that lacked Atg7 in terms of deficiencies in this process, which had a negative effect on the course of the disease.
This is a significant result since increasing age is an important risk factor in the progressive phase of MS. The researchers also show how this process can be reversed.
“The plant and fungi-derived sugar Trehalose restores the functional breakdown of myelin residues, stops the progression and leads to recovery from MS-like disease.” says doctoral student Rasmus Berglund.
“By enhancing this process we hope one day to be able to treat and prevent age-related aspects of neuroinflammatory conditions.”
Trehalose (O-α,-D-glucopyranosyl-[1 → 1]-α-D-glucopyranoside) is a disaccharide comprised of an α, α-1,1-glucosidic bond between two α-glucose units (Fig. 1a).
It is a non-reducing stable sugar, which is not readily hydrolyzed by acid or α-glucosidase. Its inert characteristic ensures that it does not readily interact with proteins or other biomolecules1,2.
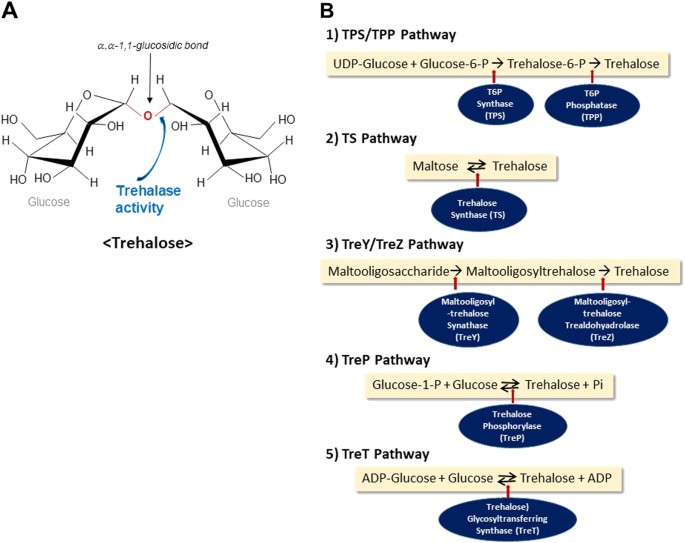
a Structure of trehalose. Trehalose consists of two glucose units linked through α, α-1,1-glucosidic bond. It is a stable non-reducing sugar, which is readily hydrolyzed by the enzyme trehalase. b Trehalose synthetic pathways. Five pathways to synthesize trehalose are shown. A most common pathway is the (1)‘‘TPS/TPP’’ pathway to form trehalose-6-phosphate, which is dephosphorylated to become trehalose. (2) Trehalose synthase (TS) synthesize trehalose from maltose. (3) Maltooligosaccharides are broken down to from trehalose by the TreY/TreZ pathway. (4) Trehalose phosphorylase (TreP) utilizes glucose-1-phosphate to form trehalose. (5) ADP-glucose is used to from trehalose by trehalose glycosyltransferring synthase (TreT).
Trehalose is detected in most organisms except for vertebrates3.
Not one gene involved in trehalose biosynthesis nor storage is found in vertebrate genomes3.
Why do vertebrates not synthesize trehalose?
Rather than losing the ability to produce trehalose in the evolution process, it seems that they never acquired such capacity in the first place. Vertebrates and invertebrates have strikingly divergent ancestors and follow separate lines in the early steps of evolution.
Most invertebrates come from protostomes, whereas vertebrates and some invertebrates, such as Echinodermata, are originated from deuterostomes. Deuterium-derived primitive organisms also do not own trehalose-synthesizing genes3.
Prominent features of trehalose arise from its non-reducing property, which leads to high hydrophilicity, chemical stability, and strong resistance to acid hydrolysis and cleavage by glucosidases. Furthermore, trehalose was shown to act as a molecular chaperone to help refold partially denatured proteins4,5.
Recent reports of trehalose as an autophagy inducer and a protector against pathological changes in various models of neurodegenerative disorders suggested this disaccharide as an attractable therapeutic option. This review provides a careful assessment of these studies and discusses the potential mechanism of neuroprotection by trehalose.
Structure and biochemical characteristics of trehalose
The chemical stability of trehalose arises from the 1,1-glycosidic linkage, which has low energy (1 kcal/mol) compared to other similar disaccharide sucrose (27 kcal/mol). It is not readily hydrolyzed into glucose units unless the enzyme trehalase is present6.
The glycosidic bond in trehalose has greater flexibility than in other disaccharides, and it facilitates the sugar to conform with other polar groups of biomolecules easily2. Trehalose has the highest ability for hydration compared to other sugars. As a result, it may enhance stabilization of membrane lipids by arranging the water molecules nearby or by direct interaction with polar biomolecules in replacement of water molecules7,8.
There are three suggested mechanisms by which trehalose stabilizes proteins: water replacement, glass transition, and chemical stability2. Trehalose inhibits protein denaturation by the exclusion of water molecules from the surface of proteins when cells are in the dehydrated condition9.
In the dry state, it maintains proteins in the folded state by replacing water molecules and forming hydrogen bonds directly with proteins10. The unique property of trehalose to create a stable non-hygroscopic glass at high temperatures in the dry state also allows maintenance of the protein structure11.
The amount of trehalose accumulation in different yeast species is related to the ability to survive heat and dehydration12,13. In nematodes, trehalose accumulates at the onset of dehydration14. Trehalose is rapidly broken down once the stress is relieved, bringing it down to the normal level.
Trehalose, therefore, acts as a natural stabilizer of life processes, withstanding extreme temperatures, nutrient deprivation, osmotic pressures, and dehydration in many species of invertebrates1,12,13,15. Trehalose serves as an excellent desiccant for many organisms. Even human primary fibroblasts, artificially producing trehalose, could be maintained in the dry state for up to 5 days16.
Trehalose in vertebrates
Trehalose metabolism
Vertebrates do not synthesize or store trehalose, but retain active hydrolyzing enzyme, trehalase, in the small intestine15. Trehalase resides in specific locations, such as intestinal mucosa and renal brush-border membranes, liver, and possibly blood50. Vertebrates express the enzyme during gestation stage. The highest concentration is reached after weaning.
The levels of trehalase from birth remains throughout adult life51. Intestinal trehalase is responsible for rapid degradation of ingested trehalose. Various organisms that constitute the human diet, including plants and fungi, contain trehalose. Like in lactose intolerance, having a low concentration of trehalase causes malabsorption, diarrhea, or other gastrointestinal symptoms52.
Intake of probiotic Saccharomyces boulardii by such patients had increased trehalase activity in the intestine and reduced those symptoms53.
Urinary trehalase has been proposed to be a specific marker for kidney damages. In diabetes, higher trehalase activity and genetic variations in the trehalase gene were noted54,55. Human trehalase (TREH) has a remarkable feature shared with yeast acid trehalase 1 (ATH1)56.
TREH rescued phenotypes of yeast ATH1 mutant but not NTH1 or NTH2. ATH1 is present in the vacuole and catalyze the hydrolysis of extracellular trehalose57,58. These results suggest that human trehalase gene TREH may act as a stress-response gene involved in the utilization of exogenous trehalose56.
Trehalose in neuroprotection and protein aggregation
Saccharides in glycoproteins and glycolipids play essential roles in the brain. They assist in brain development, synaptogenesis, synaptic transmission, and neurotransmitter production59,60. Moreover, several saccharides enhanced brain functions in animal and human studies. Dietary polysaccharides derived from yeast and plants improved cognitive function and mood in healthy young and middle-aged human adults61,62,63.
Neuroprotective properties of trehalose were mentioned in animal studies. Growing C. elegans in trehalose-containing growth medium extended the lifespan64. Mouse models with neurologic defects partially recovered from their behavioral and neurobiological defects65.
Oral administration of trehalose improved motor dysfunction and extended the lifespan of a mouse transgenic (tg) model of Huntington disease (HD)66. Superoxide dismutase 1 (SOD1) mutant tg mice had a significantly prolonged lifespan and enhanced neuronal survival with trehalose administration67. Parkin-/-/TauVLW mice had shown significant reductions in the phosphorylated tau-positive neuritic plaques and astrogliosis in the brain68.
In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease (PD), trehalose inhibited the reduction in the striatal dopamine levels and prevented gliosis69.
Evidence for trehalose as an autophagy activator and an inhibitor of protein aggregation
The initial observations of trehalose as a neuroprotective reagent in human and animal studies led to the following in vitro and in vivo studies. In a yeast study, it was first described as a potential inhibitor for the aggregation of denatured proteins4. Not only did trehalose directly stabilize proteins in the native state but it also reduced aggregation of proteins that have already been denatured.
In HD tg mouse model, trehalose decreased polyglutamine aggregates in cerebrum and liver66. In vitro aggregation of Aβ peptides was also inhibited in the presence of trehalose70. When trehalose was orally administered to SOD1 mutant tg mice, there was a decrease in the accumulation of SOD1 aggregates in the brain67.
Meanwhile, several reports proposed that trehalose might induce autophagy71. Autophagy is a lysosome-mediated degradation process to remove damaged cellular components. These include damaged organelles, such as mitochondria, endoplasmic reticulum (ER), and peroxisomes, as well as misfolded or aggregated proteins and intracellular pathogens72.
Three major types of autophagy have been described so far, which are macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy73.
Impairment of autophagy is linked to many diseases including cancer, inflammatory diseases, and neurodegenerative disorders. Environmental stresses, such as starvation, growth factor depletion, and oxidative stress, activate macroautophagy through inhibition of mTOR.
Such stresses lead to transcriptional activation of autophagy genes and downstream activation of autophagosome synthesis (Fig. 2).
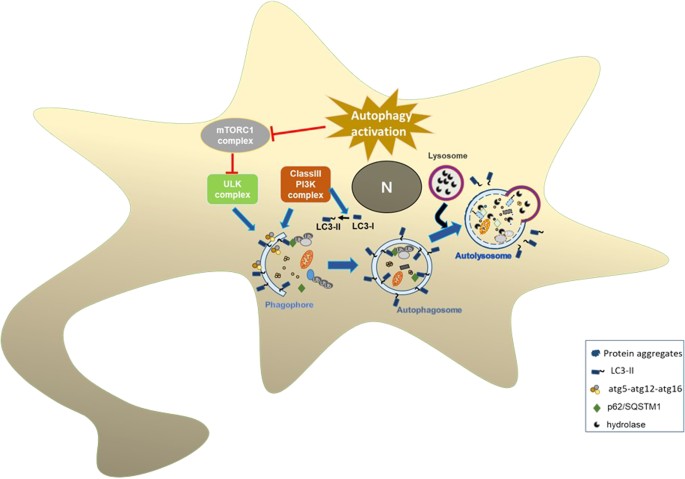
Activation of autophagy inhibits mTORC1 complex that leads to autophagosome formation. Atg5–atg12–atg16 complex helps elongation of phagophore membrane. Cytosolic LC3-I is converted to lipidated LC3-II and binds to inner and outer membranes of autophagosomes. Mature autophagosomes are formed and are ready for fusion with lysosomes. Lysosomal hydrolases degrade autophagosome contents.
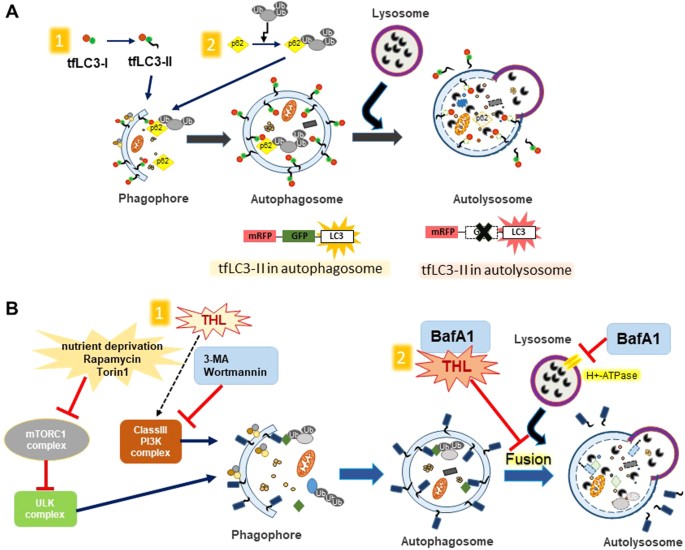
a Distinguishing between autophagosomes and autolysosomes. mRFP-GFP tandem fluorescent-tagged LC3 (tfLC3) fluoresces both GFP and RFP signals (yellow) before it is delivered to lysosomes. GFP in tfLC3 loses its fluorescence in the acidic and degradative lysosomal environment (red). Autophagy induction increases autophagosomes (yellow) and autolysosomes (red) together because the autophagic flux to lysosomes is not disturbed. Blocking fusion of autophagosomes and lysosomes, however, would increase the number of autophagosomes (yellow) only. b Autophagy modulating factors. Autophagy is initiated through inhibition of mTORC1 complex and activation of Class III PI3K complex. 3-methyladenine (3-MA) and Wortmannin prevent autophagy through inhibition of mTORC1 complex. Trehalose may activate autophagy through PI3K. BafA1 and trehalose could both inhibit fusions of autophagosomes and lysosomes, thus blocking final stage of autophagy
Bafilomycin A1 (BafA1), a vacuolar-type H+ ATPase inhibitor, is often used to inhibit the late stage of autophagy process where autophagosomes fuse with lysosomes. LC3-II levels increase with BafA1 treatment. Chloroquine, a lysosomotropic agent, also prevents autophagosome–lysosome fusion and inhibits autophagy.
A study investigated the effect of BafA1 on trehalose-modified autophagy71. COS-7 cells, transfected with Htt proteins, were treated with trehalose (100 mM, 48 h) and BafA1 (200 nM, 4 h). The results showed that LC3-II levels increased more when BafA1 and trehalose were treated together than each reagent treated alone.
In a different study, ScN2a neuronal cells treated with trehalose (100 mM, 48 h) and BafA1 (200 nM, 4 h) showed similar results that the LC3-II levels were increased more with treating both reagents together than with treating each alone78. These results were main evidence used as evidence for trehalose being an autophagy activator.
However, the use of BafA1 as an autophagy blocker raised a concern when treated for a short time ( < 6 h), which might be insufficient to block autophagy entirely82.
Temporal examination of BafA1 showed that short-term treatments with BafA1 resulted in impaired acidification of lysosomes, but only after treatments with extended periods prevented the fusion between lysosomes and autophagosomes. Therefore, it was recommended that BafA1 should be treated for an extended period ( > 6 h for most cell types) to inhibit autophagic flux. In our recent work, we have also performed similar experiments but treated BafA1 for a longer time (12 h or more)80.
The results showed that the effect of trehalose and BafA1 together was similar to BafA1alone, suggesting the role of trehalose as an inhibitor of autophagic flux.
Recent studies on trehalose aimed to uncover molecular pathways of autophagy activation and neuroprotection were reported. Trehalose prevented glucose/fructose uptake by inhibition of solute carrier 2A (SLC2A) proteins and reduced hepatic steatosis83. Impairment of glucose uptake has the same effect as the starvation, which affects autophagy.
Little is known as to how cells take up trehalose. SLC2A8 (GLUT8) was presented as a mammalian trehalose transporter, partly responsible for uptake of trehalose and trehalose-induced autophagy in hepatocytes84. However, trehalose-induced LC3-II accumulation in N2A neuroblastoma cells was not SLC2A8-dependent, because SLC2A8 is not localized in the plasma membrane in neuronal cells. These studies suggest that trehalose uptake mechanism may be different in neuronal cells compared to hepatocytes and may act differently on the autophagic pathway.
In summary, unlike the observations in animal models where trehalose administration seems to induce autophagy in the brain, trehalose seemed to be more of an autophagic flux blocker than of an autophagy inducer when treated directly to cultured cells, including neuronal cells80. tfLC3 staining and immunoblotting of autophagic markers, LC3-II and p62/SQSTM1, support the conclusion that trehalose may impair lysosomal activity and interfere with degradation through autolysosomes, acting similarly to lysosomotropic inhibitors (Fig. 3b).
Trehalose effects on other pathways
Autophagy has received the most attention as the key player in the mechanism of action of trehalose in mammalian cells. However, there have been several studies suggesting its roles in other cellular processes. Trehalose downregulated PARP-1 and PARP-2 expression after lipopolysaccharide (LPS) and interferon gamma (INFγ)-induced oxidative stress in primary rat astrocyte and oligodendrocyte cultures, suggesting the anti-apoptotic function of trehalose under oxidative stress conditions85.
Trehalose intake by Lewy body disease model mice increased levels of several chaperone molecules, such as HSP90 and SigmaR1, suggesting its roles in protein folding76. Oxygen-glucose deprivation (OGD) inhibits proteasome activity via suppression of both oxidative stress and ER stress. Trehalose inhibited OGD-induced autophagy while preserving proteasome activity86.
Trehalose may also regulate stress granules, which are RNA–protein complexes in the cytoplasm of eukaryotic cells, produced under specific stress conditions. The function of these stress granules is thought to protect RNAs from degradation, thereby preventing cell death and stress signaling.
The stress granule formation is initiated by recruitment of eukaryotic initiation factor 2 (eIF2) to form eIF2-GTP-tRNAiMet ternary complex. Many proteins in the stress granules are dysregulated in human diseases, such as ALS.
Prolonged accumulation of the stress granules may lead to increased protein aggregation and the pathogenesis of neurodegenerative diseases. Trehalose efficiently promoted the stress granule disassembly via the p-eIF2α pathway, suggesting that neuroprotective effects of trehalose may include the regulation of the stress granules87.
Does trehalose directly affect clearance of protein aggregates?
Neuroprotection and autophagic induction by trehalose led to a hypothesis that trehalose promotes aggregate clearance through activation of autophagy. Although several studies attempted to link the two processes together, the evidence provided has not been conclusive. In this section, we will critically assess the literature on this subject and examine whether trehalose can directly promote aggregate clearance.
Sarkar et al.71 reported first that trehalose has an autophagy-inducing effect and enhanced clearance of protein aggregates, such as mutant huntingtin and α−synuclein in cultured cells. A similar observations were made where trehalose treatment reduced polyubiquitinated protein aggregates that were induced by epoxomicin75.
The same study also showed the reduced tau and α−synuclein aggregates in NB69 neuroblastoma cells. In these studies, however, the quantitative analysis of the aggregates might have been limited. For example, α−synuclein immunoblotting only displayed the reduction of monomers. The high molecular weight α−synuclein aggregates were not shown.
In the latter study, the amounts of phosphorylated tau were not normalized to the total tau levels (tau-5 antibody), and both phosphorylated and total tau seems to change in similar patterns. In the case of polyQ proteins of the former study, however, immunoblotting showed the high molecular weight protein aggregates left in the wells of sodium dodecyl sulfate polyacrylamide gel electrophoresis gels. Further characterization of the aggregates with different methods would have strengthened the argument on the role of trehalose in aggregate clearance.
Another study by Tanaka et al.66 examined the effect of trehalose on polyglutamine aggregates in the Neuro2A mouse neuroblastoma cells expressing a GFP fused polyQ protein. Trehalose and other small molecules were delivered into cells directly by a lipid-based transient cell permeabilization method.
The result showed a reduction in the number of cells with aggregates and improvement in cell viability upon trehalose treatment. However, using an artificial cell delivery system might have allowed high levels of trehalose in cells that might not occur in vivo.
In a different study, cells stably infected with prion and treated with trehalose displayed more diffuse and dispersed patterns of PrPSc staining in the periphery of the cells compared to control cells which had compact, round inclusion stainings in the perinuclear area74. One thing that was noticeable in this study is that trehalose was treated at low concentrations (~50 μM), compared to 50–100 mM typically used in other studies.
Further characterizations of the compact/round and dispersed structures would help determine whether these structures represent protein aggregates or merely represent limited localization of PrPSc to specific organelles of the cell.
Trehalose treatment for longer incubation period (up to six passages) did not modify proteinase K (PK) resistance of PrPSc aggregates in this study, suggesting that trehalose may not affect protein aggregation at low concentrations.
Interestingly, several recent works reported the results that contradicted the role of trehalose in aggregate clearance. Partially denatured proteins after heat shock were efficiently refolded in the presence of trehalose, thereby suppressing aggregation in yeast4. However, the continued presence of trehalose interfered with protein refolding in yeast.
A study with amyloid precursor protein (APP) showed that trehalose decreased degradation of APP and other long-lived proteins in cells81. These effects are associated with diminished lysosomal hydrolase activities, such as cathepsin D. In our latest study, addition of trehalose into the culture medium of SH-SY5Y cells increased α-synuclein aggregation and lysosomal integrity was also impaired80.
Curiously, there was little correlation between protein aggregation and cell toxicity when trehalose was present, suggesting that the aggregates formed under trehalose treatment conditions were not toxic or that trehalose protected cells from aggregate toxicity. Mouse primary cortical neurons exposed to pre-formed fibrils (PFF) of α-synuclein had shown an increased abundance of phosphorylated S129 form and reduced cell viability88.
Trehalose failed to remove α-synuclein aggregates in these cells. However, it increased basal cell viability compared to non-treated cells. These results suggest that protective effects of trehalose may act independently from its effects on protein aggregation.
Direct vs. indirect mechanism of neuroprotection by trehalose
Studies have shown that administration of trehalose is neuroprotective in animal models. When trehalose was administered, animals with neurodegenerative diseases lived longer with reduced neuropathology and alleviated behavioral phenotypes. These animal studies also exhibited autophagy activation and the reduction of protein aggregates.
Inflammatory responses decreased, and gliosis diminished in response to trehalose. More neurons survived in the specific areas of the brain in the disease models.
In cells, however, the connection of autophagy activation and the reduction of protein aggregates to trehalose is still controversial. Careful analysis of autophagy flux and protein aggregates suggested that unlike the previous hypothesis, trehalose interferes with autophagy flux and increases, rather than decreases, the levels of protein aggregates in cultured cells.
These results raised the possibility that the cause of autophagy induction and aggregate clearance in animal models of neurodegenerative diseases may not stem from the direct effects of trehalose on neurons. The neuroprotective effects of trehalose in animals may be indirect.
What might be the mechanism of neuroprotection by trehalose? Contrary to the direct uptake of trehalose in culture, trehalose treated to animals in drinking water is likely to be hydrolyzed by trehalase enzyme in the gut. Even if some trehalose enters the blood stream, there is the blood–brain barrier (BBB) that limits the access of trehalose to the brain.
One possibility is that the effects of trehalose are exerted at the gut level. For example, trehalose may influence gut microbiota. Trehalose can protect microbes from harmful stresses and enhance the survival.
Increasing body of evidence has accumulated over the years that gut microbiota has wide-spread effects on many physiological systems, including the central nervous system, raising the possibility that trehalose exerts its neuroprotective roles through microbiota-gut-brain signaling89.
Consistent with this hypothesis, only the oral intake of trehalose, not an intraperitoneal injection, efficiently induced autophagy in the mouse brain, suggesting that the neuroprotective effects of trehalose require the gastrointestinal (GI) system76.
On the other hand, one cannot rule out the possibility that trehalose travels through the blood stream and enter the brain, exerting its neuroprotective functions directly to neurons. There are reports of trehalose detection in blood plasma and its relation to diabetes54,55. It is also detected in liver and kidney, but the functions of trehalose in these organs are still unknown. A recent study by Martano et al.90 detected trehalose in mouse brain, particularly in the hippocampus and cortex.
Endogenous trehalose was detected in both astrocytes and neurons, but the hydrolyzing enzyme, trehalase, was localized in only neurons. Astrocytes were able to take up and release trehalose into the extracellular space. The source of trehalose, however, was unclear as trehalose-synthesizing enzymes are not present in vertebrates.
These results raise a possibility that trehalose may travel from gut to brain, through blood or other carriers, and act directly on neurons. Once trehalose reaches the brain, it may act directly on neurons and other cells to affect protein folding, act as a signaling molecule to activate stress responses, or regulate autophagy and cell death mechanisms. However, it is doubtful that the concentrations of trehalose in neurons and other brain cells reach the levels at which most of the in vitro studies have been done (Fig. 4).
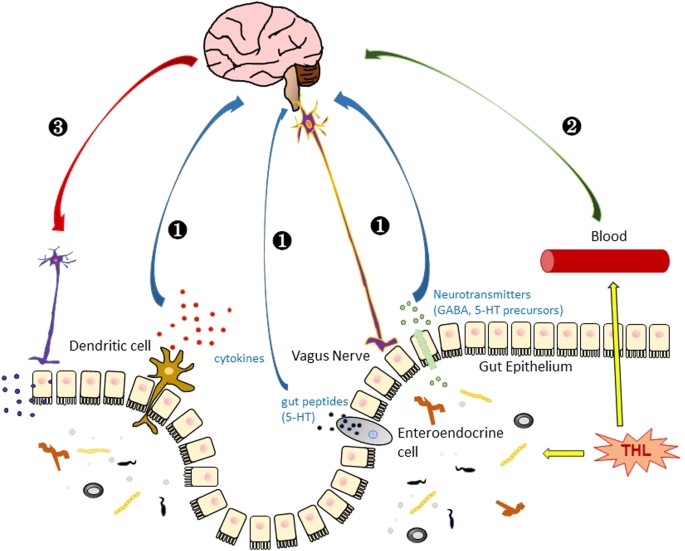
(1) Trehalose indirectly affects brain function through the regulation of gut microbes, which sends signals to the brain by dendritic immune activation or secretion of neurotransmitters and gut peptides that may be delivered through vagus nerve to the brain91. (2) Direct transport of trehalose to the brain, which passes through the blood–brain barrier and affects neuronal cells. (3) The brain sends signals to the enteric system to modulate trehalose function
reference link : https://www.nature.com/articles/s41419-018-0749-9
More information: Rasmus Berglund et al, Microglial autophagy-associated phagocytosis is essential for recovery from neuroinflammation, Science Immunology, 16 October 2020, DOI: 10.1126/sciimmunol.abb5077



{kind=link}
[…] The sugar Trehalose restores the functional breakdown of myelin residues –… […]