











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



EPFL scientists have discovered a new role for bile acids: they curb appetite by entering the brain. Their findings, which were recently published in Nature Metabolism, provide new insights into the signals and mechanisms by which satiety is controlled and may have implications for treating obesity.
Our brain is usually well protected from uncontrolled influx of molecules from the periphery thanks to the blood-brain barrier, a physical seal of cells lining the blood vessel walls. The hypothalamus, however, is a notable exception to this rule.
Characterized by “leaky” blood vessels, this region, located at the base of the brain, is exposed to a variety of circulating bioactive molecules. This anatomical feature also determines its function as a rheostat involved in the coordination of energy sensing and feeding behavior.
Several hormones and nutrients are known to influence the feeding neurocircuit in the hypothalamus. Classic examples are leptin and insulin, both involved in informing the brain of available energy. In the last years, the list of appetite- or satiety-triggering signals has been steadily growing with the identification of several gut hormones.
Those are involved in fine-tuning feeding behavior by regulating the perception of hunger or satiety, ultimately leading to the initiation or termination of a meal. The gut-brain axis is thus a critical gatekeeper in regulating feeding behavior.
Bile acids are among the most abundant metabolites in the gut and act as versatile signaling molecules that relay nutrient availability to a physiological response by activating the bile acid responsive membrane receptor, Takeda G-coupled receptor 5 (TGR5). Although the ancient Greeks already postulated that bile may affect our state of mind, we know very little about the signaling role of these metabolites in the brain.
In a new study from the Schoonjans’ lab at EPFL, together with the EPFL Brain Mind Institute and Bertarelli Platform for Gene Therapy, and several collaborators from France, Italy and the US, the authors showed that bile acids reach the mouse brain shortly after a meal to suppress food intake.
The authors demonstrated that the anorexic response of bile acids is mediated by TGR5, located at the cell surface of a distinct group of hypothalamic cells, called AgRP/NPY neurons. When focusing on this neuronal subpopulation, they found that bile acids mediate two processes staggered in time.
“While bile acids acutely block the release of appetite-stimulating AgRP and NPY peptides during the first minutes following binding of their cognate receptor, they further reinforce the repression by blunting the expression of these neurotransmitters,” says Alessia Perino, first-author of the paper.
Over the last two decades, bile acids have been proven to be efficacious in alleviating chronic metabolic and inflammatory disorders. Previous studies from the Schoonjans lab demonstrated that systemic TGR5 activation attenuates obesity in diet-induced obese mice.
The current study reveals that the bile acid-TGR5 signaling axis is not only important in disease, but also in the physiological control of eating behavior. In the absence of dietary fat, bile acids temporarily suppress food intake without affecting the normal energy balance.
“This is not surprising as homeostasis is about a self-regulatory process in which systems tend to maintain stability” says Kristina Schoonjans. “In contrast, chronic high fat diet feeding may override this equilibrium. It will be interesting to find out whether the identified neurocircuits contribute to the known body weight reducing effect of bile acids in the setting of diet-induced obesity.”
Bile acids are synthesized in the liver, where cholesterol is converted via 7α-hydroxylase (CYP7A1) and, to a lesser extent, 27α-hydroxylase (CYP27A1) and 24-hydroxylase (CYP46A1), to the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA) in humans (CA and muricholic acid in rodents).
These are then conjugated to glycine or taurine, prior to their secretion into bile [1]. Following meal ingestion, bile acids are released into the gut upon gallbladder emptying, and about 95% of intestinal bile acids is absorbed in the ileum via the apical sodium bile acid co-transporter (ASBT), returning to the liver for re-secretion—a highly efficient process known as “enterohepatic circulation”.
A small fraction of bile acids reach the large intestine, where they are modified (through de-conjugation and dihydroxylation) by gut bacteria to secondary bile acids such as deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA, a secondary bile acid in humans, but a primary bile acid in rodents), and absorbed passively into the circulation or excreted in the feces [2] (Figure 1).
Bile acids lost to the large intestine are replenished by de novo hepatic synthesis, which is regulated by fibroblast growth factor-19 (FGF19) signaling in the small intestine in humans (or FGF15 in rodents). Thus, bile acids are found in high concentrations in the liver [3], bile [4], and small intestine [5].

For more than a century, bile acids have been regarded solely as “intestinal detergents” that emulsify dietary fat for digestion and transport. The recognition that bile acids are also pivotal signaling molecules orchestrating glucose, lipid and energy metabolism is recent. Bile acids also bind to numerous nuclear and cytoplasmic receptors such as the vitamin D receptor [6], pregnane X receptor [7], and constitutive androstane receptor [8].
However, it was the identification of the bile acid-specific nuclear farnesoid X receptor (FXR) in 1999 and membrane Takeda G-protein-coupled receptor 5 (TGR5) in 2002 that provided a mechanistic framework for a role of BA signaling in the context of metabolism [9,10]. FXR and TGR5 are present in numerous tissues including the central and peripheral nervous systems; bile acid signaling in the latter has been shown to regulate energy intake [11], as supported by the observation that suppression of energy intake induced by intravenous injection of DCA is attenuated when TGR5 was silenced in the vagal nodose ganglia in rats [12].
However, the clinical relevance of this concept is unclear, particularly given that plasma bile acid concentrations are low and that in obese individuals, relative elevation in plasma bile acid levels are not associated with reduced energy intake. In line with the high turnover of bile acids in the enterohepatic circulation, both FXR and TGR5 are expressed abundantly in the liver and the intestine. Signaling through both receptors has been linked to the secretion of gastrointestinal hormones, known to be integral to the maintenance of metabolic homeostasis (Figure 1).
For example, the release of ghrelin from gastric G-cells during fasting appears pivotal to sensations of hunger, and stimulation of energy intake. After meals, the secretion of cholecystokinin (CCK) from enteroendocrine I-cells located in the upper gut, and glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) from L-cells located most abundantly in the distal gut, form an integrated signaling system that slows gastrointestinal motility and transit, drives the secretion of insulin to regulate postprandial glucose metabolism (via GLP-1) and suppresses appetite and energy intake [13].
The role of bile acids in the control of blood glucose and lipid metabolism has been reviewed in detail [14,15,16,17], but their potential to impact on the regulation of energy intake has received less attention, despite the recognition, since 1968, that oral administration of CDCA and DCA stimulated PYY secretion and suppressed appetite in obese individuals [18]. The current review addresses the effects of bile acids on gastrointestinal hormone secretion, energy intake, and body weight as well as the relevance of bile acid dysregulation in obesity and type 2 diabetes (T2D).
Effects of Bile Acids on Gastrointestinal Hormone Secretion
The last two decades have witnessed a substantial effort to increase the understanding of the effects of bile acids on gastrointestinal hormone secretion and the consequent impact on metabolism. In healthy individuals, postprandial plasma bile acid concentrations have been reported to correlate negatively with ghrelin, and positively with GLP-1 and PYY [19]. Similar relationships have also been observed in obese patients following bariatric surgery [20].
However, bile acids per se do not appear to affect ghrelin secretion in rats; intestinal infusion of a mixture of physiological bile acids did not affect portal ghrelin levels [21]. In contrast, small intestinal sensing of bile acids has been reported to inhibit CCK secretion in both rodents and humans [22,23], supporting the existence of a negative feedback loop between the two.
In contrast, the effects on GLP-1 and PYY release from L-cells have been studied extensively in preclinical and clinical models [24,25,26], stimulating the potential development of bile acid-based interventions for metabolic disorders. While bile acid-induced release of GLP-1 and PYY has been linked to signaling via FXR and TGR5, the data are inconsistent, which may relate to differences in the binding affinity of individual bile acids at FXR and TGR5 (Table 1) and/or complex interactions between the two signaling pathways.
Table 1. Binding affinities of bile acids to human TGR5 and FXR.
| Bile Acid | TGR5 | FXR | ||||
|---|---|---|---|---|---|---|
| Subjects | Indicator | EC50 | Subjects | Indicator | EC50 | |
| Primary Bile Acids | ||||||
| CA | CHO cells/HEK293 | Intracellular cAMP | 7.72 µM [34]/ >10 µM [10] | CV-1 cells | Reporter gene activation | No effect [45] |
| CDCA | CHO cells/HEK293 | Intracellular cAMP | 4.43 µM [34]/ 4 µM [10] | HepG2 cells /CV-1 cells | Reporter gene activation | 10 µM [9]/ 50 µM [45] |
| CHO cells | Reporter gene activation | 6.71 µM [46] | Cell-free | Ligand-sensing assay | 4.5 µM [47] | |
| Conjugated Primary Bile Acids | ||||||
| TCA/GCA | CHO cells | Reporter gene activation | 4.95 µM/ 13.6 µM [46] | Cell-free | Ligand-sensing assay | No effect [47] |
| TCDCA/ GCDCA | CHO cells | Reporter gene activation | 1.92 µM/ 3.88 µM [46] | Cell-free | Ligand-sensing assay | 10 µM [47] |
| HCA | Cell-free | TR-FRET FXR coactivator assay | 70.06 µM (IC50) [28] | |||
| Secondary bile acids | ||||||
| DCA | CHO cells | Intracellular cAMP | 1.01 µM [34] | HepG2 cells | Reporter gene activation | 100 µM [9] |
| HEK293 | Intracellular cAMP | 575 nM [10] | CV-1 cells | Reporter gene activation | 50 µM [45] | |
| LCA | CHO cells | Intracellular cAMP | 0.53 µM [34] | CV-1 cells | Reporter gene activation | 50 µM [45] |
| HEK293 | Intracellular cAMP | 35 nM [10] | Cell-free | Ligand-sensing assay | 25 µM [6] | |
| UDCA | CHO cells | Reporter gene activation/Intracellular cAMP | 36.4 µM [46]/ No effect [34] | CV-1 cells | Reporter gene activation | No effect [45] |
| HDCA | CHO cells | Reporter gene activation | 31.6 µM [46] | Cell-free | TR-FRET FXR coactivator assay | 62.43 µM [28] (IC50) |
| Conjugated Secondary Bile Acids | ||||||
| TDCA/ GDCA | CHO cells | Reporter gene activation | 0.79 µM /1.18 µM [46] | Cell-free | Ligand-sensing assay | 500 µM [47] (IC50) |
| TLCA/ GLCA | CHO cells | Reporter gene activation | 0.29 µM /0.54 µM [46] | Cell-free | Ligand-sensing assay | 3.8 µM/4.7 µM [47] (IC50) |
| TUDCA/ GUDCA | CHO cells | Reporter gene activation | 30.0 µM /33.9 µM [46] | Cell-free | Ligand-sensing assay | No effect [47] |
| THDCA/GHDCA | CHO cells | Reporter gene activation | 24.2 µM/36.7 µM [46] |
Note: EC50: the concentration for a half maximal effect; IC50: the concentration for a half maximal inhibitory effect; CHO: Chinese hamster ovary cells; HepG2 cells: Human hepatoma cell line; CV-1 cells: Monkey kidney fibroblast cells (CV-1 line); HEK293: human embryonic kidney cell line 293; TR-FRET FXR coactivator assay: commercial assay kit for screening ligand for FXR.
FXR
FXR is expressed abundantly in the liver and the intestine, and the binding affinity of individual bile acids is variable (CDCA > DCA > LCA > CA > UDCA, Table 1). FXR was initially identified as a regulator of bile acid metabolism [14], and subsequently as a modulator of L-cell secretion. Indeed, FXR is expressed by the murine L-cell line, GLUTag. However, the FXR agonist GW4064 and CDCA (which preferentially binds FXR) were shown to suppress glucose-induced proglucagon expression and GLP-1 secretion in this cell line by decreasing glycolysis, whereas silencing FXR abolished these effects [27].
These observations have been replicated in studies with different L-cell lines (i.e., NCI-H736 [28] and STC-1 [29]). In a similar manner, GW4064 blunted the GLP-1 response to short-chain fatty acids (SCFA) in both GLUTag and NCI-H716 cell lines [30]. Consistent with these observations, FXR-deficient mice exhibited increased GLP-1 secretion in response to both dietary fiber, which increases colonic SCFA [30], and oral glucose [31]. Oral intake of GW4064 (10 mg/kg, 2 doses over 12 h) also decreased active GLP-1 levels in the plasma of mini-pigs [28].
However, in an isolated perfusion model of rat intestine, both luminal and vascular perfusion of GW4064 failed to affect the GLP-1 response to a physiological mixture of bile acids in rats [21]. In mice, diversion of bile acids from the gallbladder to the ileum was shown to modestly increase GLP-1 secretion, improve glucose tolerance, and induce weight loss [32]. The reductions in postprandial blood glucose and body weight induced by this procedure were abolished in intestinal FXR-knockout mice, suggesting that intestinal FXR-signaling can potentially promote GLP-1 secretion.
Unfortunately, the study failed to determine whether the rise in GLP-1 was specifically induced by FXR-activation [32]. Of note, oral administration of the intestine-restricted FXR agonist, fexaramine, in mice was reported to increase the abundance of LCA-producing gut bacteria to activate TGR5-signaling indirectly, leading to enhanced GLP-1 secretion and improvement in insulin sensitivity and lipid profile as well as the promotion of adipose tissue browning [33]. Accordingly, outcomes derived from ex vivo and in vivo experiments are, by and large, inconsistent, although the intestine-restricted FXR signaling appears to have an overall favorable effect on metabolic health.
TGR5
TGR5, also known as GPBAR1, is a G-protein coupled receptor that is expressed widely in the gastrointestinal tract, pancreas, liver, gallbladder, and adipose tissue. Like FXR, its binding affinity for individual bile acids varies substantially (LCA > DCA > CDCA > CA > UDCA, Table 1) [34]. TGR5 activation has been reported to suppress hepatic macrophages, induce gallbladder relaxation and refilling, and promote intestinal motility [14].
TGR5 is also expressed on L-cells. Unlike FXR, stimulation of TGR5 by LCA and DCA was shown to potently stimulate GLP-1 secretion from STC-1 cells in a dose-dependent manner, an effect suppressed by downregulation of TGR5 expression [35]. The stimulatory effect of TGR5 on GLP-1 secretion required the closure of ATP-sensitive potassium (KATP) channels and elevated intracellular concentrations of cAMP and Ca2+ [36,37].
A major observation in relation to TGR5 signaling was the demonstration of its basolateral location on L-cells. Thus, to activate TGR5, it is necessary for bile acids or other TGR5 ligands to be transported through the epithelial layer [38]. However, the readily absorbed TGR5 agonist SB-756050 failed to stimulate GLP-1 secretion significantly, or improve glycemic control at various doses compared with the placebo in acute studies involving patients with T2D [39]. It is noteworthy that L-cells are distributed most densely in the distal gut regions [13]. It would therefore be of interest to investigate whether delivery of TGR5 agonists should be targeted at the distal gut.
PYY is co-released with GLP-1 from L-cells, and it was initially noted that perfusion of DCA (1–25 mM) into the isolated rabbit colon increased PYY secretion substantially in a dose-dependent manner [18]. Intracolonic administration of DCA or TCA in humans has also been shown to induce a rapid and substantial rise in plasma PYY [40,41,42]. Similar to TGR5-mediated GLP-1 secretion, the outcomes of studies using isolated rat colon indicate that bile acid-induced PYY secretion is dependent on bile acid translocation from the luminal to basolateral side [43]. That PYY secretion is less evident in response to bile acids with poor affinity to TGR5, and attenuated in TGR5-knockout models, attests to the fundamental relevance of TGR5-signaling to bile acid-induced PYY secretion [44].
In summary, there is compelling evidence for a role of bile acids in the modulation of GLP-1 and PYY secretion in both animals and humans. Stimulation of TGR5 on L-cells induces the secretion of both hormones, while effects of FXR signaling remain controversial. The interactions between FXR and TGR5 signaling remain poorly characterized and an improved understanding may be of relevance to the development of novel strategies for the management of metabolic disorders.
Effects of Bile Acid Signaling on Energy Intake and Body Weight
In light of the effects of bile acids on appetite regulation, particularly via the secretion of gastrointestinal hormones, it is intuitively likely that modulating bile acid signaling affects energy balance. Genetic ablation of the bile acid synthesis enzyme CYP8B1, leading to a deficiency of 12α-hydroxylated bile acids (e.g., CA), has been shown to be associated with reduced energy intake and subsequent weight gain in mice fed a fat enriched diet [48,49]. However, these effects appeared to be secondary to impaired fat hydrolysis and the increased exposure of unabsorbed fat to the distal gut, as in these mice, there was an increase in energy intake when fed a fat-free diet [49].
Nevertheless, this study supports the fundamental role of endogenous bile acids in fat digestion and absorption, which may influence energy intake and body weight indirectly.
The outcomes of preclinical and clinical studies involving administration of various bile acids have been equivocal in relation to effects on energy intake and body weight (Table 2). For example, supplementation with CA or UDCA prevented weight gain in mice fed a high-fat diet [50,51,52], possibly reflecting a TGR5-related increase in energy expenditure [50]. Moreover, a number of other bile acid species with high affinity for TGR5 including hyocholic acid (HCA), hyodeoxycholic acid (HDCA), DCA, and TCA failed to affect energy intake or body weight in rodents with or without diabetes [28,53,54].
Information relating to the effects of bile acids on appetite and energy intake in humans are limited. In healthy individuals, rectal administration of TCA substantially stimulated GLP-1 and PYY secretion and suppressed hunger in a dose-dependent manner [42]. Similarly, in obese individuals with T2D, rectally administered TCA significantly suppressed energy intake dose-dependently [41].
However, these observations could be confounded by the concurrent urge for defecation induced by rectal TCA perfusion (Figure 2) [42]. More recently, a double-blind, randomized, placebo-controlled 4-week trial that delivered a mixture of encapsulated bile acids (1000mg/day) designed for release in the ileum and colon (to provide dual agonism of FXR and TGR5) showed little effect on body weight in patients with T2D, despite increases in plasma GLP-1 and serum and intestinal bile acids [55].
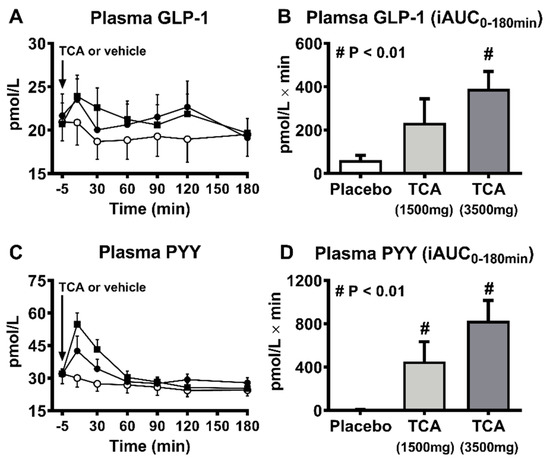
Table 2. Reported effects of bile acids on energy intake and body weight in preclinical and clinical models.
| Bile Acid | Model | Dose | Method | Effect | Ref | |
|---|---|---|---|---|---|---|
| Conjugated Bile Acid | ||||||
| Primary | TCA | HFD Sprague-Dawley rat + streptozotocin | 0.05% or 0.3% | Fed with high-fat diet for 12 weeks | Body weight − Energy intake − | [54] |
| Patients with T2DM | 0.66, 2, 6.66, or 20 mmol | Rectal administration | Energy intake ↓ (~47% at 20 mmol) | [41] | ||
| HCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] | |
| TUDCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] | |
| Secondary | HDCA | db/db mice; HFD C57BL/6J mice + streptozotocin; C57BL/6J mice | 100 mg/kg/day | Oral gavage for 28 days | Body weight − Energy intake − | [28] |
| Unconjugated Bile Acid | ||||||
| Primary | CA | C57BL/6J mice | 0.5% | High-fat diet fed for 47 days | Body weight ↓ (24%) Energy expenditure ↑ (~50%) Energy intake − | [50] |
| 0.5% | High-fat diet fed for 9 weeks | Body weight ↓ (6g, ~18%) Energy expenditure ↑ (29%) Energy intake ↑ (20%) | [52] | |||
| UDCA | 0.5% | High-fat diet fed for 8 weeks | Body weight ↓ (15%) | [51] | ||
| Secondary | DCA | C57BL/6J mice | 0.1% | High-fat diet fed for 3 weeks | Body weight − Energy intake − | [53] |
Note: EC50: the concentration for a half maximal effect; IC50: the concentration for a half maximal inhibitory effect; CHO: Chinese hamster ovary cells; HepG2 cells: Human hepatoma cell line; CV-1 cells: Monkey kidney fibroblast cells (CV-1 line); HEK293: human embryonic kidney cell line 293; TR-FRET FXR coactivator assay: commercial assay kit for screening ligand for FXR. Cholic acid (CA); Chenodeoxycholic acid (CDCA); Taurocholic acid (TCA); Glycocholic acid (GCA); Taurochenodeoxycholic acid (TCDCA); Glycochenodeoxycholic acid (GCDCA); Hyocholic acid (HCA); Deoxycholic acid (DCA); Lithocholic acid (LCA); Ursodeoxycholic acid (UDCA); Hyodeoxycholic acid (HDCA); Taurodeoxycholic acid (TDCA); Glycodeoxycholic acid (GDCA); Taurolithocholic acid (TLCA); Glycolithocholic acid (GLCA); Tauroursodeoxycholic acid (TUDCA); Glycoursodeoxycholic acid (GUDCA); Taurohyodeoxycholic acid (THDCA); Glycohyodeoxycholic acid (GHDCA).
As discussed, physiological bile acids often activate both FXR and TGR5, but with preferential affinity depending on their molecular structure. Selective FXR- and TGR5-knockout mice, or specific FXR and TGR agonists, have been pivotal to delineation of the respective signaling pathways to the metabolic effects of bile acids. However, outcomes remain inconclusive. Administration of the intestinal FXR agonist, fexaramine, for five weeks to mice fed a high-fat-diet was reported to prevent weight gain.
However, this may have reflected an increase in metabolic rate, rather than a reduction in energy intake [56]. In contrast, GW4064 had no effect on either energy intake or body weight in diabetic or obese mice [50,57]. Notably, mice with FXR deficiency (either whole body or intestine-specific knockout) fed a high-fat diet also exhibited reductions in energy intake and body weight compared with wild-type mice [31,58].
Similarly, TGR5 agonism (e.g., by INT-777) was associated with reduced weight gain, apparently by augmenting energy expenditure, without affecting energy intake [36], whereas knockout of TGR5 had no significant effect on body weight or energy intake in mice fed a high-fat diet [36,59]. Clinical outcomes relating to TGR5 or FXR agonism have been disappointing. As discussed, the TGR5 agonist, SB-756050, failed to stimulate GLP-1 secretion or improve glycemic control in individuals with T2D [39].
The effects of TGR5 agonists on energy intake and body weight in humans have not been reported. Treatment with the semi-synthetic FXR agonist, obeticholic acid, over 72 weeks only achieved a modest reduction in body weight (~2 kg) in patients with non-alcoholic fatty liver disease (NAFLD), with or without, T2D [60]. In another 24-week double-blind, randomized, placebo-controlled trial, the non-bile acid FXR agonist, cilofexor, had no effect on body weight in patients with non-alcoholic steatohepatitis [61]. Accordingly, the concept of supplementing bile acids or targeting BA signaling pathways to reduce energy intake and body weight is currently not supported by current clinical evidence.
Bile Acid Dysregulation in Obesity and T2D
The emerging link between bile acid signaling and the regulation of metabolic homeostasis has stimulated substantial interest in potential phenotypical changes in bile acid profiles in metabolic disorders, particularly obesity and T2D. Although bile acids are present at high concentrations in the liver, bile, and small intestine, bile acid profiles have hitherto been compared in peripheral blood and fecal samples predominantly due to their easy accessibility. Accordingly, processes in relation to small intestinal bile acid transport and absorption are poorly characterized, although gallbladder emptying can be readily assessed using ultrasound.
There is a substantial variation in circulating bile acid levels both between and within individuals [62]. In the context of obesity, most studies have reported that fasting serum/plasma bile acid levels are increased as a result of augmented bile acid synthesis (reflected by an increase in 7α-hydroxy-4-cholesten-3-one (C4)) [63,64,65]. There is evidence that the expression of both hepatic Na+-taurocholate co-transporting polypeptide (NTCP) [66] (responsible for the uptake of bile acids from the portal vein to the liver) and intestinal ASBT is lower in obese individuals [67], and intestinal FGF-19 secretion is also decreased [67,68].
It is, therefore, conceivable that the augmented hepatic bile acid secretion observed during fasting represents a compensatory response to deficiencies in the enterohepatic circulation. In support of this concept, the postprandial increase in circulating bile acids is significantly blunted in obesity [66,69,70] and restored after Roux-en-Y gastric bypass [70]. In addition, the production and fecal excretion of secondary bile acids (e.g., DCA) are increased in obese individuals [71,72], which may be secondary, or contribute to, alterations in gut microbiota (“dysbiosis”) [73], leading to impaired energy metabolism in the host [74].
Obesity-related increases in fasting bile acid levels primarily reflect increases in 12α-hydroxylated bile acids (e.g., CA) [64,66], which are more effective in emulsifying dietary fat than non-12α-hydroxylated bile acids [49]. The shift in the bile acid composition in obesity may, therefore, favor improved fat digestion. Although fasting plasma unconjugated primary bile acids (CA and CDCA) and numerous conjugated primary and secondary bile acids (TCA, GCA, GCDCA, TDCA, and GLCA) are related positively with insulin resistance in obesity [75,76], it remains to be determined whether changes in plasma bile acids represent a manifestation, or the drivers, of obesity.
T2D individuals, with or without obesity, exhibit higher fasting bile acid concentrations in the peripheral circulation compared with non-diabetic controls, mainly due to increased unconjugated and glycine-conjugated DCA and UDCA [64,77,78,79,80]. This rise in plasma secondary bile acids may reflect increased bile acid delivery and a relative abundance of bile acid de-conjugating bacteria in the large intestine [81,82].
Interestingly, the expression of ASBT has been reported to be increased in diabetic rats [83], which would favor enhanced ileal bile acid resorption. However, this does not necessarily lead to increased FGF-19 secretion [77,79,80], or suppression of bile acid synthesis in T2D. Hepatic bile acid synthesis, particularly CA, is, in fact, known to be increased in patients with T2D [80]. In a small group of individuals with T2D (n = 15), the plasma BA responses to oral glucose or fat-containing mixed nutrients were reported to be modestly elevated [77].
Gallbladder emptying in this group of patients was similar to healthy controls [84]. However, in this study, T2D patients had relatively poor glycemic control (mean HbA1c = 7.5%) and a long duration of diabetes (6–20 years), with the majority receiving medication (e.g., metformin [85]) known to affect bile acid metabolism.
The magnitude of the increase in fasting bile acids in plasma or serum has been shown to correlate positively with fasting and 2 h-postprandial glucose levels and HbA1c in T2D, and with the degree of insulin resistance in individuals, regardless of the presence of diabetes [79,86].
In a recently reported longitudinal study, 23 bile acid species were analyzed to evaluate their baseline association with incident T2D during a median 3-year follow-up in a large cohort of individuals with normal glucose tolerance [87]. Serum fasting unconjugated primary and secondary bile acids (CA, CDCA, and DCA) were reported to be negatively associated with the risk of T2D, while conjugated primary and secondary bile acids (GCA, TCA, GCDCA, TCDCA, and TUDCA) were positively associated.
Moreover, the ratios of conjugated to unconjugated bile acids (TCA/CA, GCA/CA, TCDCA/CDCA, and GCDCA/CA) were positively associated with the development of T2D. These observations support the concept that impaired catalysis of conjugated bile acids by the hepatic bile acid-CoA:amino acid N-acyltransferase (BAAT) [88] and/or intestinal resorption of unconjugated bile acids contribute to the development of T2D.
The relevance of postprandial bile acid levels, particularly in the small intestine and liver, to the risk of T2D, however, remains unknown. Further studies are, therefore, required to clarify how bile acid metabolism changes with the progression of glucose dysregulation.
reference link: https://www.mdpi.com/2072-6643/13/4/1104/htm
More information: Central anorexigenic actions of bile acids are mediated by TGR5, Nature Metabolism (2021). DOI: 10.1038/s42255-021-00398-4



{kind=link}