")










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Variante B.1.1.519 : I ricercatori avvertono che la variante messicana B.1.1.519, che sta devastando il Messico da alcuni mesi, non solo provoca un aumento del rischio di ospedalizzazione e gravità della malattia, ma aumenta anche il rischio di mortalità.
Inoltre, proprio come le varianti Delta e Mu, sta generando sottovarianti che destano preoccupazione.
Sono già stati pubblicati due studi da ricercatori messicani, uno che descrive in dettaglio l’emergere della variante e l’altro che descrive il suo panorama evolutivo e il suo impatto clinico a Città del Messico.
https://link.springer.com/article/10.1007/s00705-021-05208-6
https://www.medrxiv.org/content/10.1101/2021.09.07.21262911v2
Nel primo studio, il team di ricerca ha riportato l’identificazione di una potenziale variante di interesse, che ospita le mutazioni T478K, P681H e T732A nella proteina spike, all’interno del nuovo lignaggio B.1.1.519 che ha rapidamente superato le varianti preesistenti in Messico ed è stato il virus dominante nel paese durante il primo trimestre 2021.
L’analisi complessiva del genoma dei virus nel lignaggio B.1.1.519 ha mostrato la presenza di 20 mutazioni in totale, rispetto alla sequenza del genoma di riferimento Wuhan-Hu-1 (numero di accesso NCBI MN908947). Undici di queste mutazioni non sono sinonime e quattro di esse sono presenti nella proteina spike. https://link.springer.com/article/10.1007/s00705-021-05208-6/tables/1
La variante B.1.1.519 è raggruppata in un clade indipendente derivato dalla classificazione clade 20B NextClade secondo l’analisi filogenetica. La variante B.1.1.519 è caratterizzata da nove mutazioni, quattro delle quali sono sostituzioni ORF1 e tre sostituzioni spike.
In particolare, una mutazione T478K è presente nel dominio di legame del recettore (RBD), dove è stato dimostrato che le mutazioni riducono l’attività di alcuni anticorpi monoclonali. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8053237/
Il lignaggio B.1.1.519, rappresentato dalla stragrande maggioranza delle sequenze messicane riportate, è stato identificato per la prima volta come un lignaggio B.1.1.222 . Tuttavia, la presenza delle mutazioni T478K, P681H e T732A lo ha chiaramente differenziato da questo ceppo, che non contiene queste mutazioni, dando origine al ceppo B.1.1.519.
Un’analisi in silico utilizzando diverse strutture potenti di ceppi correlati ha suggerito che la posizione della mutazione T478K nella proteina S è coinvolta nel riconoscimento dell’anticorpo e nel sito di legame del recettore.
In una scansione mutazionale profonda del dominio di legame del recettore SARS-CoV-2, la mutazione T478K non ha avuto un effetto significativo sul ripiegamento o sul legame con l’enzima di conversione dell’angiotensina umana 2 (ACE2). https://pubmed.ncbi.nlm.nih.gov/32841599/
Si ipotizza tuttavia che questa mutazione possa essere coinvolta nell’evasione immunitaria, in particolare nella neutralizzazione dell’anticorpo. https://pubmed.n cbi.nlm.nih.gov/33535027/
La mutazione P681H è una delle mutazioni riscontrate anche nella variante B.1.1.7 rilevata nel Regno Unito. È stato scoperto che aumenta la scissione degli spike da parte di proteasi simili alla furina e alcuni ipotizzano che potrebbe aumentare la trasmissibilità.
La mutazione T732A e la delezione 69-70 sono state trovate anche nella proteina spike. Si ritiene che entrambi svolgano un ruolo nell’evasione immunitaria.
Nel secondo studio, il team di ricerca ha scoperto che la variante messicana B.1.1.519 con le mutazioni P681H, T478K e T732A era responsabile del 90% dei casi rilevati nel febbraio 2021 a Città del Messico.
Il team ha riportato il numero effettivo di riproduzione di B.1.1.519 e ne presenta l’origine geografica sulla base dell’analisi filogenetica. È stata anche studiata l’evoluzione di questa variante attraverso l’analisi degli aplotipi e sono stati identificati gli aplotipi più recenti. Il team ha anche esaminato l’impatto clinico dei pazienti infetti dalla variante B.1.1.519 rispetto agli individui infetti da SARS-CoV-2 non B.1.1.519.
Secondo lo studio, il 3 novembre 2020 è stato identificato il primo paziente infetto dalla variante B.1.1.519 a Città del Messico, solo il secondo caso registrato in tutto il mondo.
Tuttavia, all’interno di Città del Messico, la frequenza delle varianti è aumentata dal 16% al 90% nel febbraio 2021, ma è diminuita al 51% nel maggio 2021.
È stato riscontrato che delle sequenze generate da infezioni da SARS-CoV-2, la variante B.1.1.519 rappresentava 74,3 % da novembre 2020 a maggio 2021 a Città del Messico. La variante B.1.1.519 è stata rilevata in 31 paesi, costituendo il 55% dei casi in Messico.
Per determinare la trasmissibilità della variante B.1.1.519, il gruppo di ricerca ha studiato il numero di riproduzione efficace (Rt), che è stato definito come il numero medio di casi secondari per caso primario in un dato momento entro l’anno. È stato osservato un forte aumento della Rt per la variante B.1.1.519, fino a un valore di 2,9 nella seconda settimana di dicembre 2020, che corrispondeva all’aumento della rilevazione. Tuttavia, il valore di Rt ha iniziato a stabilizzarsi nei mesi successivi, tra 0,5 e 1.
È importante sottolineare che la variante B.1.1.519 è stata la seconda più frequentemente rilevata a Città del Messico. Il valore di Rt per la variante B.1.1.519 ha oscillato fortemente nei mesi successivi a dicembre, il che potrebbe essere stato influenzato dal piccolo numero di casi associati a questa variante.
Questa forte fluttuazione differisce da altre varianti rilevate in quella regione poiché altre varianti si sono stabilizzate o sono scomparse nel corso dell’anno.
È stata calcolata la filogenesi massima dettagliata, che includeva tutti i genomi SARS-CoV-2 di interesse per esaminare l’origine geografica della variante B.1.1.519 e la sua relazione evolutiva con la variante B.1.1.222.
Ci sono tre cluster definiti mostrati nell’albero filogenetico. Due corrispondono solo alle varianti B.1.1.519 e B.1.1.222, con separazione netta e cluster misto, che mostra una separazione indefinita tra i lignaggi.
Ciò dimostra che l’evoluzione di questo lignaggio della variante SARS-CoV-2 rimane poco chiara, sebbene il cluster misto formato dalle sequenze della variante B.1.1.519 sia più strettamente correlato alle sequenze della variante B.1.1.222.
Il team di ricerca ha studiato l’impatto clinico della variante B.1.1.519 analizzando le associazioni tra la variante e il numero di tratti clinici. Le uniche sequenze considerate per le analisi sono state quelle con set completi di dati clinici (N=600).
La valutazione allarmante dei dati ha rivelato che i pazienti infetti dalla variante B.1.1.519 hanno mostrato un aumento significativo della probabilità di sviluppare sintomi che interessano il tratto respiratorio rispetto alle varianti non B.1.1.519.
Modelli dettagliati di regressione logistica aggiustati per soglia del ciclo virale, numero di comorbidità, età e sesso hanno rivelato che la variante B.1.1.519 era associata a un aumento di 1,786 volte della mancanza di respiro, un aumento di 1,489 volte del dolore toracico e un Aumento di 3,655 volte della cianosi. I
noltre, la variante B.1.1.519 è stata associata a una frazione più elevata di pazienti che hanno sviluppato malattie gravi o morte.
Il team di studio ha osservato che la variante B.1.1.519 SAR-CoV-2 era significativamente associata a ospedalizzazione, malattia grave e morte. I pazienti infettati con la variante B.1.1.519 sembravano anche mostrare una maggiore prevalenza di sintomi come mancanza di respiro, dolori al petto e cianosi.
Va notato che gli esiti più gravi associati alla variante B.1.1.519 sono simili a quelli mostrati con la variante delta. Recenti ricerche hanno dimostrato che le infezioni con la variante delta mettono i pazienti a un rischio più elevato di malattie gravi che portano al ricovero in ospedale.
Nonostante i rapporti inviati all’OMS, l’agenzia sanitaria internazionale, piuttosto lenta e compiacente, non ha ancora elevato la variante B.1.1.519 a quella di Variant of Concern o VOC!
I ricercatori messicani avvertono che la variante si sta ancora diffondendo a livello globale e, peggio ancora, vengono trovate nuove sottovarianti del B.1.1.591 con mutazioni e delezioni uniche e alcuni stanno preoccupando i team di ricerca.
Entrambi i team di studio avvertono che la sorveglianza genomica sostenuta è fondamentale per identificare nuove varianti emergenti. Eventuali associazioni cliniche significative potrebbero essere di grande importanza per tentare di contenere la pandemia.
In America Latina, ad eccezione di P.1 e P.2 osservate in Brasile, finora non sono state segnalate altre varianti con potenziale di rapida espansione [11]. Qui, riportiamo l’identificazione di un potenziale VOI che ospita le mutazioni T478K, P681H e T732A nella proteina spike, all’interno del nuovo lignaggio B.1.1.519, derivato dal lignaggio B.1.1.222, che ha rapidamente superato il preesistente varianti in Messico ed è stato il virus dominante nel paese durante il 2021.
Derivate dalla sorveglianza genomica effettuata in Messico, in questo studio sono state ottenute 2.692 sequenze genomiche e fanno parte delle 3.156 sequenze depositate nel GISAID dal 1 marzo 2020 al 21 marzo 2021. Come risultato dell’analisi di questo insieme di sequenze , abbiamo osservato la presenza di 91 linee di assegnazione filogenetica di Named Global Outbreak (PANGO), con B.1.1.519 (37,8%), B.1 (13,9%), B.1.1.222 (10,3%), B.1.1 (5,7%), B.1.609 (5,6%) e B.1.243 (4,5%) sono i più diffusi.
Le librerie per il sequenziamento dell’intero genoma di SARS-CoV-2 sono state generate utilizzando il protocollo sviluppato dalla rete ARTIC (https://artic.network/2-protocols.html) o un metodo basato su lunghi ampliconi (https://pubmed .ncbi.nlm.nih.gov/32222995/).
Un’osservazione sorprendente è stata la scoperta del ceppo B.1.1.519 negli Stati Uniti, derivato da B.1.1.222, che ospita la mutazione T478K nella proteina spike. Questa variante non era stata rilevata in Messico prima dell’ottobre 2020, quando è stata trovata a Città del Messico, e l’analisi filogeografica ha suggerito che la variante B.1.1.519 è emersa intorno alla metà di settembre 2020 [10].
A novembre 2020, il 13% (16/123) dei casi caratterizzati di COVID-19 era causato da questa variante e a dicembre questa percentuale è salita al 29,3% (97/331). A gennaio 2021 la percentuale di B.1.1.519 è salita al 51,5% (229/445), aumentando di incidenza al 73,6% (808/1098) a febbraio. È stata invece osservata una frequenza decrescente del ceppo B.1 che aveva predominato in Messico nel 2020, passando dal 36,27% (284/783) tra marzo e settembre al 2,37% (26/1098) nel febbraio 2021.
In Messico, dall’identificazione di B.1.1.519 nel novembre 2020, sono state segnalate un totale di 6419 sequenze genomiche con questo lignaggio. La maggior parte di loro proviene da Città del Messico ed è diffusa in tutto il paese.
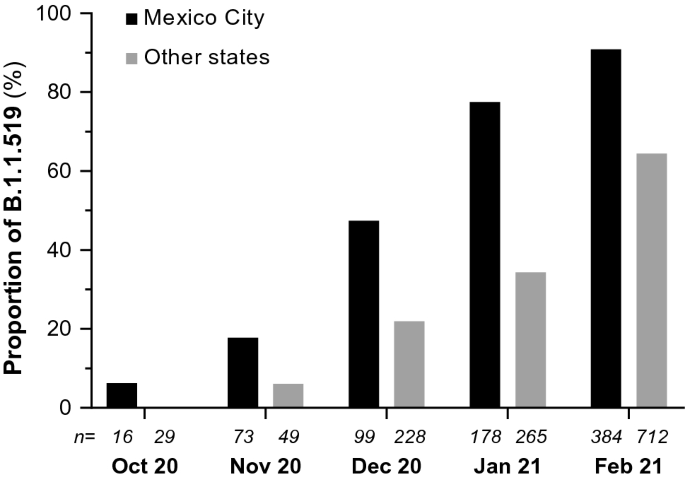
Un’analisi dettagliata dei campioni provenienti da Città del Messico ha indicato che, a novembre, questa variante era presente nel 17,8% (13/73) dei casi, mentre a dicembre 2020 questa percentuale è salita al 47,5% (47/99). A gennaio 2021 la variante è stata rilevata nel 77,5% (138/178) dei casi e a febbraio nel 90,9% (349/384).
Questo significativo aumento della frequenza di B.1.1.519 a Città del Messico ha mostrato che ha superato le varianti preesistenti tra ottobre 2020 e febbraio 2021 e questo aumento è stato osservato anche in altre regioni del paese (Fig. 1), che rappresentano più di 50 % dei virus caratterizzati in alcuni stati durante il primo trimestre del 2021.
In particolare, la variante era molto diffusa in Baja California Sur (51,3%, 20/39), Guerrero (70%, 21/30), Hidalgo (72,2% , 13/18), Morelos (67,3%, 33/49), Stato del Messico (83,5%, 76/91), Oaxaca (51.
Questa variante è stata rilevata anche in 17 paesi di tutti e cinque i continenti. Nelle Americhe, è stato segnalato in Canada e negli Stati Uniti, e recentemente in Brasile, Cile, Aruba, Martinica e Curazao [12]. Tuttavia, questa variante attualmente non è predominante in questi paesi.
L’analisi complessiva del genoma dei virus nel lignaggio B.1.1.519 ha mostrato la presenza di 20 mutazioni in totale, rispetto alla sequenza del genoma di riferimento Wuhan-Hu-1 (numero di accesso NCBI MN908947). Undici di queste mutazioni non sono sinonime e quattro di esse sono presenti nella proteina spike.
In particolare, una mutazione T478K è presente nel dominio di legame del recettore (RBD), dove è stato dimostrato che le mutazioni riducono l’attività di alcuni anticorpi monoclonali [9]. Tutti i cambiamenti di aminoacidi e nucleotidi sono elencati nella Tabella 1.
Tabella 1 Cambiamenti di aminoacidi e nucleotidi
| mutazioni | Gene | |
|---|---|---|
| nucleotide | Amminoacido | |
| C203T | – | |
| C222T | – | |
| C241T | – | |
| C3037T | – | ORF1a |
| C3140T | P141S | |
| C10029T | T492I | |
| C10954T | – | |
| A11117G | I49V | |
| C12789T | ||
| C14408T | P323L | ORF1b |
| T19839C | – | |
| C21306T | – | |
| C22995A | T478K | Arpione |
| A23403G | D614G | |
| C23604A | P681H | |
| A23756G | T732A | |
| G28881A | – | n |
| G28882A | R203K | |
| G28883C | G204R | |
| C29197T | – |
- Le principali sostituzioni di amminoacidi sono mostrate in grassetto
L’attuale linea B.1.1.519, rappresentata dalla stragrande maggioranza delle sequenze messicane riportate, è stata identificata per la prima volta come linea B.1.1.222. Tuttavia, la presenza delle mutazioni T478K, P681H e T732A lo ha chiaramente differenziato da questo lignaggio, che non contiene queste mutazioni, dando origine al lignaggio B.1.1.519. Un’analisi filogenomica delle sequenze genomiche utilizzando lo strumento Nextstrain ha mostrato che i virus nel gruppo della stirpe B.1.1.519 (B.1.1.1.222+T478K+P681H+T732A) indipendentemente dalle sequenze della stirpe B.1.1.222, suggerendo fortemente che questa variante dovrebbe essere classificata come variante di interesse (VOI) (Fig. 2). D’altra parte, i virus della linea B.1.1.519 sono già raggruppati dalla piattaforma GISAID in un clade indipendente, che ospita invariabilmente le tre mutazioni sopra menzionate.
Un’analisi in silico utilizzando diverse potenti strutture di ceppi correlati ha suggerito che la posizione della mutazione T478K nella proteina S è coinvolta nel riconoscimento dell’anticorpo e nel sito di legame del recettore [13]. In una scansione mutazionale profonda del dominio di legame del recettore SARS-CoV-2, la mutazione T478K non ha avuto un effetto significativo sul ripiegamento o sul legame all’enzima di conversione dell’angiotensina umana 2 (ACE2) [14].
Tuttavia, questa mutazione può essere coinvolta nell’evasione immunitaria, in particolare nella fuga dalla neutralizzazione anticorpale [15]. La mutazione P681H è una delle mutazioni trovate nella variante B.1.1.7 rilevata nel Regno Unito. Secondo le definizioni descritte nel documento pubblicato dall’OMS “Covid-19 Weekly Epidemiological Update” del 25 febbraio 2021, con l’edizione speciale di “Proposed Working Definitions of SARS-CoV-2 Variants of Interest and Variants of Concern”, possiamo considerare il ceppo B.1.1.519 una potenziale variante di interesse [16].
Infine, in questo studio sono state identificate due varianti con caratteristiche interessanti: in primo luogo, 13 sequenze appartenenti al lignaggio B.1.1.222 senza la mutazione T478K, ma che ospitano la mutazione T732A e la delezione 69-70 nella proteina spike, quest’ultima essendo una mutazione caratteristica del VOC B.1.1.7 rilevata per la prima volta nel Regno Unito; e in secondo luogo, 11 sequenze corrispondenti a quattro lignaggi differenti da B.1.1.519 (B.1, B.1.1.222, B.1.1.322, e B.1.323) ma contenenti le stesse mutazioni T478K, P681H e T732A in le glicoproteine spike che sono presenti nella variante B.1.1.519. Si raccomanda di tenere traccia dell’incidenza di queste due varianti durante la sorveglianza genomica.
Finora, non abbiamo prove sperimentali per determinare se le mutazioni qui descritte potrebbero essere associate a cambiamenti nella trasmissione, virulenza e/o antigenicità o se potrebbero avere un impatto sulla gravità della malattia, sui tassi di reinfezione o sull’efficacia del vaccino. Per questo motivo, l’importanza di un sistema di sorveglianza genomica, studi epidemiologici ed esperimenti per valutare la neutralizzazione dei virus nel ceppo B.1.1.519 o eventuali nuove varianti sono cruciali per indagare il possibile impatto biologico delle mutazioni nel contesto pubblico Salute. Fortunatamente, tutte le varianti del virus COVID-19 emerse finora rispondono in una certa misura ai vaccini disponibili e approvati.
collegamento di riferimento: https://link.springer.com/article/10.1007/s00705-021-05208-6



{kind=link}