











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Epigenetics is commonly defined as the study of heritable phenotypic changes without altering the DNA sequence. The Greek prefix epi- (ἐπι) in epigenetics implies a function “above” or “in addition to” the traditional genetic base [15].
Over the past two decades, epigenetic regulators have been implicated as critical factors in many pathways related to the development and progression of cancer and other diseases, including cell cycle regulation, invasiveness, signaling pathways, chemotherapy resistance, and immune evasion [16,17,18,19,20,21,22,23,24,25,26,27].
The three basic systems of epigenetic regulation are DNA methylation of gene regulatory regions; histone protein modifications such as methylation, acetylation, phosphorylation, and sumoylation; and non-coding RNAs [15]. Many techniques for epigenetics analysis have already been developed, and this field is steadily undergoing technological innovation [15,28,29].
reference link: https://www.mdpi.com/2227-9059/9/9/1142/htm
Epigenetics
Epigenetics has been described as the area of life sciences that studies stably heritable phenotype resulting from changes in chromatin structural/activation states without altering the DNA primary nucleotide sequence [32]. Currently, the epigenomics—the study into genome epigenetics—gives us the capability to read, locate, and interpret functionally the epigenetic machinery that controls the entire genome at various levels [31].
During the last decades, a large body of research provided evidence that epigenetics plays an important role in the establishment and progression of many common diseases, particularly those related to age (age-related diseases; ARD) [33]. Moreover, the patterns of gene expression determined by molecular epigenetic marks established during development affect the vulnerability to several diseases in humans, including viral infections [18].
Remarkably, epigenetic alterations involve changes in the chromatin structure or the nucleic acid chemical properties without hacking the genetic code, unlike mutations that directly derange the genetic material [32]. This characteristic makes epigenetic alterations reversible, flexible, and quickly responsive to environmental changes and other exposures [34]. Indeed, prolonged exposure to altered metabolic conditions may epigenetically affect human cells [35].
Several epigenetic mechanisms work together to regulate gene expression synchronizing the metabolic information. At the chromatin level, DNA methylation and histone modifications lead to chromatin remodeling, and together with other modifying proteins (sirtuins, prions, etc.) and non-coding RNA (miRNA, sRNA, lncRNA), allow chromatin access to proteins that regulate DNA transcription and, therefore, RNA and protein synthesis [36, 37].
The cell epigenome reflects the gene activation state of chromatin by encoding the information about how and where gene-specific activation switches are located and used in the genome [38]. Chromatin is a complex of proteins and DNA; the nucleosome, composed of two copies of four core histones (H3, H4, H2A, and H2B), is the fundamental unit. The DNA wraps around the histone octamer, which, thanks to histones specific chemical composition, regulates DNA access for gene transcription [39].
Chromatin remodeling controls many epigenetic processes through a series of dynamic changes in the structural organization of nucleosome by reversible histone and DNA modifications, which result in different chromatin condensation levels [40]. Among the hundreds of enzymes involved in the epigenetic regulation of relevance for this article are histone acetyltransferases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and kinases (HKs), all acting directly on structural chromatin components.
While others, including DNA methyltransferase enzymes (DNMTs), ten-eleven translocation proteins (TETs), and Thymine DNA glycosylase (TDG), are involved in the active DNA methylation/demethylation process. All these enzymes are responsible for the establishment of specific patterns that generate affinity for chromatin-associated proteins leading to their synergistic or antagonistic interaction, and resulting in the dynamic transitions between transcriptionally active or silent chromatin states, contributing to the cellular developmental plasticity and in particular contexts, leading to pathological outcomes [33, 40].
Moreover, ncRNAs – the transcribed part of the genome that lacks protein-coding potential [41] – play a significant role in post-transcriptional and gene expression regulation by establishing or mediating the epigenetic processes, for instance, by silencing or activating genes/transcripts through different mechanisms of action. ncRNAs can modulate cell behavior and have also been implicated as inter-cellular messengers [42].
Of note, depending on the subcellular localization, lncRNAs can mimic transcription factor binding sites, acting as a decoy, or can bind to exon/intron junctions of pre-mRNA and influence the splicing process [43]. Recently, researchers provide evidence about modifications in RNA molecules such as the methylation of adenine in position 1 (N1-methyladenosine, m1A) and 6 (N6-methyladenosine, m6A) performed by the N6-adenosine methyltransferase-like 3 (METTL3) [44,45,46].
In particular, mRNAs stability is altered when m6A occurs at their 5′-AGG (m6) AC-3′ consensus sequence, affecting their translation efficiency. Moreover, as for 5-methylcytosine (5mC), the m6A of RNA can be oxidized into N6-hydroximethyladenosine (hm6A) and N6-formyladenosine (f6A) and therefore demethylated, which might lead to a different RNA–protein interaction altering gene regulation. At the RNA level, these processes are catalyzed by the fat mass and obesity-associated protein (FTO) [47].
Noteworthy, RNA and DNA methyltransferases, such as members of the NOP2/Sun domain family and the DNA methyltransferase type 2 (DNMT2), respectively, can methylate ribocytidines at position 5 (5mC) [48, 49]. Interestingly, TET enzymes act similarly on DNA and RNA molecules, converting RNA 5mC into 5-hydroxymethylcytosine (5hmC), facilitating the translation of RNA molecules [50]. Likewise, methyl groups can also be transferred at position 7 of the riboguanosine (7-methylguanosine; m7G) [51].
This modification usually occurs on capped and recapped mRNAs and is mediated by canonical mRNA capping methyltransferase (RNMT), regulating mRNA translation into proteins [52]. Of note, thanks to the modern next-generation sequencing techniques, it was possible to create high-resolution epigenome maps of healthy and diseased cells, enabling simultaneous analysis of genetic and epigenetic changes, genome-wide genetic association studies (GWAS), and epigenome-wide association studies (EWAS), respectively [53].
Epigenetic landscape alteration by viral infection
Tracking the epigenetic changes in pathophysiological contexts might represent an exciting source of knowledge to develop novel treatments leading, for example, to the regulation of the host immune response [54]. Most of the viruses belonging to the family of corona and influenza virus are usually incapable of hacking the host genetic sequence, while they might alter the host epigenome. Recent research has focused on how viruses utilize aspects of the epigenetic machinery to enable the infection establishment, spread, and persistence [55].
Furthermore, thanks to the recent advancements in high throughput technology, it is now possible to evaluate the epigenetic landscape at a genome-wide scale. It has been demonstrated that several viruses’ families antagonize the immune system by employing a series of epigenetic mechanisms, and, likely, the SARS CoV-2 might use the same strategy.
Besides, several reports highlighted how viruses might disrupt the epigenetic network regulation impacting on the host immune response. For example, Marazzi et al. have shown how the highly pathogenic H3N2 influenza A virus inhibits the initiation of the host innate immune response by interfering with the epigenetic control of gene expression [56].
Exploiting histone mimicry, it has been demonstrated that the carboxy-terminus of the H3N2 nonstructural protein NS1 shares homolog sequences with the amino-terminus of the histone H3 tail [56]. Briefly, the viral NS1 protein mimics the histone tail of the H3 histone, interacting with the transcription complex, which usually docks to the H3K4 mark to initiate transcription, interfering with the antiviral gene function.
Similarly, both the hepatitis C virus (HCV) and adenoviruses have proteins that interfere with epigenetic functions and alter global immune function [57, 58]. In 2014, Baric’s group found a clear association of repressive histone modification Histone 3-Lysine 27 trimethylation (H3K27me3) with down-regulated interferon (IFN)-stimulated genes (ISGs) following both MERS-CoV and influenza viruses A/influenza/Vietnam/1203/2004 (H5N1-VN1203) infection [59]; consequently, despite transcription factors and signaling pathways activation, the repressed state physically prevents transcription of these genes.
More recently, Menachery and coworkers also observed that DNA methylation plays a similar role in the loss of antigen-presentation molecules following MERS-CoV and H5N1-VN1203 infection. Likewise, histone methylation was found involved in abating the immune response in H5N1, through the activity of the viral protein NS1 [60]. Importantly, their sequencing data suggested that other specific regions of the genome, perhaps encoding critical genes involved in viral antagonism, are also targeted by methylation. Specifically, DNA methylation was the primary suspect in suppressing the production of antigen presentation molecules in both diseases [60].
Another study by Schäfer and Baric indicated that SARS-CoV and MERS-CoV could delay or offset pathogen recognition and ISGs expression levels by encoding unique proteins that prevent immune signaling response [18]. Based on how other viruses like human immunodeficiency virus 1 (HIV-1) and herpes modulate chromatin, they suggested that these newer viruses may also act similarly [61, 62].
Interferons are essential mediators of antiviral actions and initiators of pathogen-driven immune response by the inactivation of ISGs [63, 64], and many viruses might develop antagonistic mechanisms to overcome specific ISG effectors [65]. Indeed, during the infection, IFN and innate immune responses are extensively regulated by specific epigenetic marks, through the manipulation of the epigenetic enzyme activity and chromatin remodeling complexes formation.
The epigenetic machinery is responsible not only for the priming and the memory of host responses but also for ensuring their operational control. Fang et al. correlated the levels of Histone 3-Lysine 9 dimethylation (H3K9me2) with IFN expression in vitro. H3K9me2 is a repressive histone mark that controls the DNA methylation and heterochromatin formation processes. Specifically, the H3K9me2 mark hinders acetylation by recruiting the heterochromatin protein 1 family [66].
However, in the above study, Fang et al. demonstrated that the overall levels of H3K9me2 mark in the promoter region of the type I interferon and the expression of ISGs inversely correlate dendritic cells, defining this histone modification as an IFN response important regulator [66, 67].
On the other hand, the Histone 3-Lysine 4 trimethylation (H3K4me3) mark, commonly present in active promoters, is often enriched in Toll-like receptors (TLRs) promoter regions. Kaikkonen and coworkers have recently demonstrated that 60 min after lipopolysaccharide (LPS) stimulation of macrophages and dendritic cells, the overall histone acetylation and the binding of polymerase II (Pol II) to specific promoters was increased, suggesting a specific epigenetic regulation of the innate immune response induction [68].
Schäfer et al., using ChIP-PCR approaches, could determine differential occupancy of histone marks at the promoters of ISG genes, showing that the promoter regions of ISG genes contained more histones with active marks of H3K4 monomethylation (H3K4me) than the repressive H3K27me3 mark, therefore favoring open chromatin and promoting active transcription and ISG expression during H1N1 and SARS-CoV infection [18].
Otherwise, in MERS-CoV infected cells, Menachery et al. observed an increase in the H3K27me3 levels and reduced H3K4me3 levels at the promoter region of several specific ISGs subsets, which were not upregulated. These findings indicated that these viruses had developed antagonistic mechanisms to target the IFN innate immune response [59].
RNA type viruses, such as SARS-CoV, also show strong associations with RNA modifications. For instance, N6-methyladenosine (m6A) and N6,2′-O-dimethyladenosine (m6Am) modifications (m6A/m) have been found to play essential roles in the viral life cycle. In particular, they can affect the structure and replication of the virus, the host innate immune response, and some innate sensing pathways.
The m6A RNA methylation is the most abundant epitranscriptomic modification of eukaryotic mRNAs and has been detected on cellular and viral transcripts, regulating numerous biological processes, including viral infection [69, 70]. Imam and colleagues suggested that m6A and its associated machinery regulate the DNA virus hepatitis B (HBV) life cycle, finalized through an RNA intermediate, termed pregenomic RNA (pgRNA).
These observations indicated that m6A regulates HBV gene expression and reverse transcription. Indeed, by silencing the methylases that introduce the m6A modification to the RNA, they observed an increase in the HBV protein expression levels, while the pgRNA reverse transcription seemed reduced [71]. They mapped the m6A site in the HBV RNA and found that a conserved m6A consensus motif situated in the epsilon stem loop structure is the site for m6A modification.
This loop is located in the 3′ terminus of all HBV mRNAs and at both the 5′ and 3′ ends of the pgRNA. Immam et al. identified an m6A site in the 5′ epsilon stem loop of pgRNA by mutational analysis, revealing that m6A is required for efficient reverse transcription of pgRNA. Furthermore, their finding suggested that m6A methylation of the 3′ epsilon stem loop resulted in the HBV transcripts destabilization, indicating a double regulatory function of m6A for HBV RNA.
Whereas, Tan and coworkers provided evidence that m6A and m6Am of messenger RNA mediate diverse cellular functions by examining the viral and cellular m6A/m epitranscriptomes during Kaposi’s sarcoma-associated herpesvirus (KSHV) latent and lytic infection. KSHV transcripts are characterized by a high level of m6A/m modifications established during latent and lytic replication, conserved during the infection of different cell types [72].
Tan et al. showed that during lytic replication, upon YTH N6-methyladenosine RNA binding protein 2 (YTHDF2) knockdown, KSHV RNA degradation is impaired. YTHDF2 binds to viral transcripts and differentially mediates their stability. Moreover, they observed that KSHV latent infection-induced 5′ untranslated region (UTR) hypomethylation and 3′ UTR hypermethylation might alter the host epitranscriptome affecting the oncogenic and epithelial-mesenchymal transition processes. At the same time, KSHV lytic replication induces a dynamic reprogramming of the viral epitranscriptome itself [72].
Finally, Marz’s group observed a consistent 5mC methylation signature of coronavirus RNA. Specifically, analyzing 5mC content across various RNAs, they observed consistent methylation patterns in corresponding genomic positions of different RNAs, suggesting that the methylation of coronavirus RNAs is sequence-specific or controlled by RNA structural elements [73]. Table 1 summarizes the epigenetic implication in viral infection and their functional outcomes.
Table 1 Relevant epigenetic implication in viral infection
| Epigenetic modification | Virus infection | Target | Functional outcome |
|---|---|---|---|
| Histone methylation | H3N2 influenza A | H3K4 | Inhibition of the initiation of the host innate immune response [55] |
| SARS-CoV | H3K4me | Promotion of active transcription and ISG expression [16] | |
| H3K4me3 | |||
| H1N1 | H3K4me | Block of antiviral gene function [16, 54] | |
| MERS-CoV | H3K27me3 | Down-regulation/inactivation of ISGs [16, 57, 63] and development of antagonistic mechanism to target the IFN innate immune response [59] | |
| H3K4me3 | |||
| HSV | – | Down-regulation/inactivation of ISGs [59, 60] | |
| H5N1-Vn1203 | H3K27me3 | Down-regulation of ISGs [16, 57] | |
| HIV-1 | – | Down-regulation/inactivation of ISGs [59, 60] | |
| Histone acetylation | Adenovirus (Ad) E1A | H3K9ac | Interference with epigenetic functions and global immune function [55] |
| H3K27ac | |||
| DNA methylation | SARS-CoV | – | Delay/offset of pathogen recognition and modulation of ISG expression levels [16] |
| MERS-CoV | – | Loss of antigen-presentation molecules [58] | |
| HSV | – | Delay/offset of pathogen recognition and modulation of ISG expression levels [16] | |
| H5N1-Vn1203 | – | Loss of antigen-presentation molecules [58] | |
| HIV-1 | – | Delay/offset of pathogen recognition and modulation of ISG expression levels [16] | |
| HCV | – | Interference with global immune function [56] | |
| RNA methylation | KSHV | m6A/m6Am | Mediation of the stability of the viral transcripts [70] |
| SARS-CoV | 5mC | Modulation of the structure and the viral replication [67, 68] | |
| HBV | m6A | Regulation of gene expression and reverse transcription; transcript destabilization [69] |
Epigenetic implication in SARS- CoV-2 infection and therapy
In recent years, epigenetics evolved quickly, giving us better knowledge about inheritability functions, memory mechanisms, and developmental biology. The studies into the human epigenome are becoming more relevant in oncology, immunology, and infectious diseases [74, 75]. Indeed, during the last decade, the epigenetic research provided evidence that DNA and RNA viruses developed functions that antagonize the regulatory machine of the host epigenome by altering the host metabolism and gene expression, setting up a permissive environment for virus replication and spread [76, 77].
Furthermore, there is much evidence indicating that age-related changes to the host epigenome might compromise immune cell composition and function, affecting viral defenses, including the adaptive immune response [10, 12]. Coronaviruses, such as MERS-CoV and SARS-CoV-1, are known to mediate epigenetic alterations by antagonizing host antigen presentation or activating interferon-response genes [59, 60].
Evaluating the DNA methylation age of immune cells and other blood cell types before, during, and after infection could help explain how the aged epigenome impacts disease severity and how the virus alters the aged epigenome [10]. The vulnerability of the elderly to SARS-CoV-2 may also have to do with the effect of the epigenome on viral entry [78]. This process is initiated on the cell surface by physical interaction between the viral spike glycoprotein receptor, the ACE2 protein [26], and a co-receptor, the dipeptidyl peptidase-4 (DPP4) [30].
Nowadays, there are no specific antiviral drugs against COVID-19 infection yet, and vaccines are still under development. Even so, many potential therapeutic approaches are under investigation, and more research is urgently needed to identify effective vaccines and safe drugs for treating COVID-19 infections in order to develop pre- and post-exposure treatments against the pathogen. Although the first aim would be generating SARS-CoV-2 S-based vaccines, with conserved epitopes, able to elicit broadly neutralizing antibodies or virus-specific T cell responses, the identification, and development of safe and effective drugs to overcome SARS-CoV-2 entry and replication is essential.
Many strategies for COVID-19 treatment have been and still are under investigation: several antiviral drugs, among them Favipiravir (ClinicalTrials.gov Identifier: NCT04336904), Umifenovir (CTI: NCT04476719) or Lopinavir/Ritonavir (CTI: NCT04386876), alone or in combination with other chemicals such as the antimalarial chloroquine/hydroxychloroquine (e.g., CTI: NCT04328285); biologicals, such as convalescent plasma (e.g., CTI: NCT04321421) or mesenchymal stem cell (MSC) and MSC-derived exosomes (CTI: NCT04276987); Chinese traditional medicines (e.g., CTI: NCT04544605) and supplementation with Vitamins C and D (CTI: NCT03680274 and NCT04449718; https://www.clinicaltrials.gov/) [79,80,81,82].
Epigenetic research might help to accomplish these tasks thanks to a better understanding of the mechanisms involved in viral chromatin modification in lytic viruses and about host-virus interactions, including genetic factors that contribute to the protective or pathogenic host responses.
Clinical trials, FDA approved epigenetic-targeted agents, and combination therapy of epigenetic and antiviral drugs is currently considered as useful and beneficial for viral replication impairment and the control of the host immune response [83]. Remarkably, pharmacokinetic and pharmacodynamic properties of antivirals may also be influenced by epigenetic regulation, highlighting, once again, their relevance in the treatment SARS-CoV-2 infection [84].
Recently, El Baba and coworkers analyzed several epigenetic mechanisms involved in coronaviruses infections, identifying some major epigenetic player which can be therapeutically targeted [83]. Indeed, many of the nonstructural proteins involved in viral transcription, replication, and maturation processes are regulated by different classes of HDACs, implying that HDAC inhibitors, such as Vorinostat or suberanilohydroxamic acid (SAHA), combined with antivirals, might be useful tools to interfere with these processes [85, 86].
Of note, previous studies already showed that ACE2 expression is regulated by DNA methylation and histone modifications. In this context, epigenetic enzymes responsible for the modifications mentioned above, such as DNMT1, histone acetyltransferase 1 (HAT1), histone deacetylase 2 (HDAC2), and lysine demethylase 5B (KDM5B), become potential targets to control the host immune response [87, 88]. Therefore, DNMT1 inhibitors, e.g., Azacitidine, HAT1 inhibitors, as the anacardic acid, and HDAC2 inhibitors, as the valproic acid, may be repurposed against CoVs infections [79, 82, 89].
Moreover, knowing that viruses depend on the host epigenetic machinery, epigenetic drugs already used in cancer therapies might be exploited for their broad-spectrum antiviral action and inflammatory control [83, 90]. Indeed, some evidence indicated that the main culprit behind COVID-19 deaths is the cytokine storm, characterized by an uncontrolled over-production of soluble markers of inflammation.
Decitabine or 5-aza-2-deoxycytidine (5-azadC), a nucleoside-based DNMT inhibitor, is widely used to inhibit DNA methylation in macrophages; thus, suppressing inflammation and IFN response [83]. Noteworthy, Decitabine has recently been included in a clinical trial for COVID-19 Pneumonia-ARDS Treatment (CTI: NCT04482621).
Interestingly, the polycomb repressive complex 2 (PRC2), which mediates transcription repression via H3K27me3 enrichment at specific IFN-stimulated genes, could also be considered a target. Pharmacologic inhibitors of PRC2 are currently in advanced clinical trials for cancer treatment and could be easily repurposed to treat COVID-19 patients [91].
Recent studies show that innate immune cells may possess a form of memory, termed Trained Immunity (TRIM), a long-term boosting of innate immune response mainly maintained by natural killer cells and lung innate lymphoid cell group 2 through common epigenetic mechanisms [92, 93]. The exposure to an initial stimulus leads these cells to a metabolic, mitochondrial, and epigenetic reprogramming, which results in a memory phenotype of enhanced immune responses after the exposition to a secondary, heterologous stimulus [94].
Geller et al. also evaluate the potential effects of β-glucan about the immune dysregulation and cytokine storm observed in COVID-19. In their studies, they observed that β-glucan-driven TRIM also determines some epigenetic changes and that it could represent a useful target for COVID-19 treatment [94].
Recent studies also propose vitamins and natural products, as epigenetic modifiers, to enhance immunity and reduce the inflammatory response in COVID-19 patients [95,96,97]. For instance, the use of Vitamin D and quercetin could be interesting for ameliorating SARS-Cov-2 severity by inhibiting the expression of ACE2 and its possible role in suppressing the cytokine storm associated with mortality in COVID-19 patients [96, 98].
RNA-based drugs are other epigenetic tools that should be investigated for treating viral infections [99, 100]. For instance, among all the SARS-CoV genome that have been under study so far, Baldassarre and coworkers suggested that the 5′URT region and specific portion of it, which are essential for viral RNA replication and transcription, could be considered relevant to design novel therapeutic molecules to treat the infection [101].
Novel strategies employing small interfering RNAs (siRNAs), microRNAs (miRNAs), and locked nucleic acid antisense oligonucleotides (LNA) or GapmeRs, targeting, for instance, the 5′URT or regions of the Spike molecule, represent potential therapeutic tools for both prophylaxis and therapy of COVID-19 [101,102,103].
Indeed, the design of antisense oligonucleotides, such as Miravirsen, under investigation for HCV treatment, could be used to inhibit viral replication by scavenging miRNAs that are involved in the process [104, 105]. These studies suggest that RNA-based drugs could be optimized and employed to interfere with SARS-CoV-2 replication and transcription. Figure 2 summarizes some of the epigenetic targets and interventions potentially useful for Coronavirus viral infections treatment.
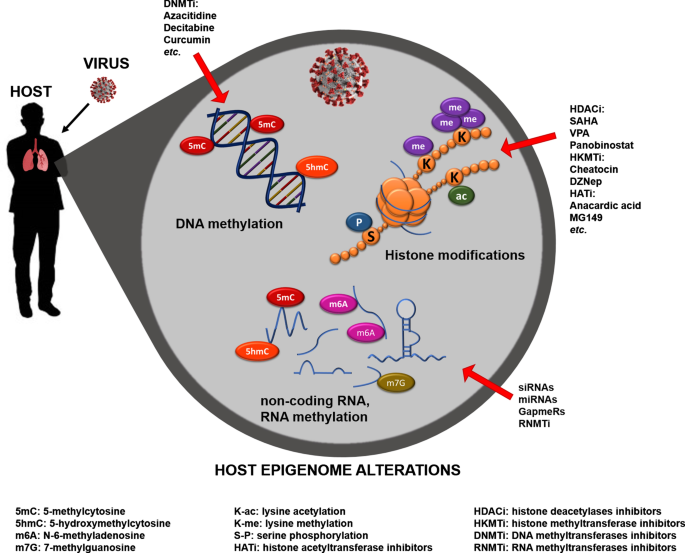
Noteworthy, thanks to sophisticated bioinformatics software, we are now able to visualize and interpret the epigenomic data, providing in-depth cell-specific knowledge about the genetic and epigenetic predispositions of an individual and explaining how the environment affects the function of our genes by leaving long-term marks on the genome. Indeed, epigenome mapping, together with EWAS and GWAS studies, provides us with tools in the diagnostics of many common human diseases, indicating that these studies could be employed for individual diagnosis and personalized therapies [106, 107].
Therefore, by investigating the epigenetic effects of metabolism from genes to pathways to genomes, and thanks to the availability of the novel detailed epigenomic datasets, we may explore how to therapeutically prevent, attenuate, or reverse the epigenetic alterations, how to design and realize specific pharmacological tools and when/where to intervene. Above all, the enzymes responsible for the epigenetic alterations represent an exciting field for discovering new drug targets.
https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-020-00946-x
Epigenetic approach of coronavirus immune evasion
Epigenetic alterations of ACE2R determines SARS-CoV entry into the host cell
Maintaining a latency stage inside the host, mimicking the host immune system requires manipulation of chromatin and heterochromatin assembly by viruses. It has become clear that the coronavirus’ spike protein facilitates its entry into the target cells mostly by the surface unit s1 of S protein upon S protein priming. The molecular affinity between the ACE2 receptor, mostly expressed on the Type–II lung epithelial cells, is crucial in the viral entry [47–50]. Spike protein and receptor affinity is a crucial determinant of tissue tropism, which plays a vital role in the disease’s etiopathogenesis. Therefore, it is crucial to understand the epigenetic signature of the ACE2 gene for controlling the initial step of entry and fusion of the virus.
The genome-wide DNA methylation array and chip methylation pipelines study indicates the varied degree of DNA methylation of the ACE2 gene in different tissue subtypes. The lowest ACE2 gene methylation across three CpG sites (cg04013915, cg08559914, cg03536816) was predominant in lung epithelial cells compared with other tissues [51]. A subsequent study shows that ACE2 gene hypo-methylations are mostly confined to the females compared with the male, suggesting angiotensin
II metabolism and its association with hormonal differences or genetic differences in chromosome dosage [52]. Moreover, transcriptomic analysis shows a lack of possible association of ACE2 with race, age and gender. However, Asian smoker population exhibits higher ACE2 than the nonsmokers suggesting an epigenetic impact on ACE2 activity in the respiratory system [53–55]. However, the validation of the approach necessitates proteomic data.
Previous evidence also highlighted hypo-methylation mediated overexpression of ACE2 and its association with the onset of severity in the patients of systemic lupus erythematosus, an autoimmune disease upon infection of SARS-CoV-2 with peripheral blood T cells [56].
Inconsistent with this, further study shows the prospective role of TNF-α in the regulation of ACE gene transcription and pathological complexity in endothelial cells. The results indicate that TNF-α enhances DNA methylation in the ACE promoter by decreasing the activity of DNMTs, DNMT3a and DNMT3b and TET1 [57]. Transcriptomic and system biology approach revealed the significant association of higher expression of ACE2 with RAB1A, HAT1, HDAC2 and KDM5B in patients with other comorbidities like hypertension, diabetes and chronic obstructive lung disease [58].
Also, the role of NAD+ dependent histone deacetylase, SIRT1 in the induction of ACE2 activity by stimulating the ACE2 promoter was reported during energy stress indicates SIRT1 could be a target for epigenetic drugs in context to COVID-19 infection [54]. Pathway enrichment analysis also revealed the potential role of KDM5B in regulating the expression of several genes associated with ACE2 possibly by acetylation and methylation epigenetic marks like H3K4me1 and H3K4me3, as well as H3K27ac [56]. Although the literature is scanty about the COVID-19 ACE2 epigenetic pattern, the molecular mechanism regulating the ACE2 activity cannot be undermined so far as the current pandemics and pathogenesis are concerned. A recent study has observed that SARS-CoV-2 cross-reactive CD4+ T cells are still present in 70% convalescent patients even after 5 months of the infection [57].
Coronavirus induces epigenetic modulation of immune cells, alters antigen presentation & interferon response
It is apparent that the generation of useful anti-inflammatory cytokines and chemokines inhibit viral replication and enhances antigen presentation [57–59]. The ISG response plays a prominent role in controlling the viral infection for efficient immune function. Type 1 IFN induces a cascade of signaling events which causes transcription of several ISG [60,61].
Although evidence is scanty from SARS-CoV-2 in this perspective, studies from influenza and other RNA respiratory viruses provide significant insight. SARS-CoV-2 and MERS-CoV viruses are found to delay ISG expression significantly. Transcriptomics and proteomics findings in Calu3 cells revealed diverse virus-specific ISG expression signatures. SARS-CoV-2 infection of Calu3 cells revealed a strong induction of ISG effectors, but the response was significantly delayed with peak expression at 48 h post-infection. In 2012, the newly emerged MERS-CoV showed a dramatic delayed ISG released with effects visible at 18 h post-infection, for the decreased expression of potential ISG subsets [62].
Further evidence supports the notion that downregulation of ISGs is not due to any impairment in the signaling cascade, but by histone modification like methylation and acetylation induced by a pathogen. Upon viral infection like SARS-CoV-2, the host produces Type I and III IFN, which induces histone modulation complex, which renders removal of repressive histone mark (H3K27me3) inducing activating mark like H3K4me3. This conversion of inactive chromatin to active chromatin allows the binding of several transcription factors like STAT1 and IRF7 thus inducing the ISG expression [63,64].
However, incorporating repressive histone modifications like H3K27me3 and removing active mark H3K4me3 could impose a more condensed state of chromatin which prevents binding of transcription factor and thus reduces ISG expression. Additionally, inhibition or downregulation of an H3K79 methylase, Dot1L enzyme associated with decreased antiviral response and facilitates viral replication, suggests its crucial role in antiviral response [65].
The previous study’s prospective interleukin role in regulating the epigenetic signature has been provided [66]. The author revealed that treatment with IL-1 induces STAT-6 activity; a major transcription factor for IL-4 mediated signaling that binds to the H3K27 demethylase Jmjd3 promoter. An elevated level of Jmjd3 decreases H3K27 dimethylation and trimethylation (H3K27me2/3), leading to transcriptional activation of the M2 marker gene. The significant association of H3K9me2 as a suppressor of IFN inducible antiviral response has been elucidated by Fang et al. (2012) [67,68].
However, inactivation of lysine methyltransferase G9a an inducer of H3K9me2 resulted in high IFN production, suggesting that methyltransferase could be an ideal therapeutic epigenetic target challenge the viral invasion. Interestingly, results of Menachery et al. (2018) revealed a possible association of H3K27me3 global methylation with downregulation of ISG and DNA methylation of antigen presentation gene upon MERS-CoV and H5N1VN1203 [66].
The possible association of TNF-α and H3K4me3 in the induction of trained innate immunity in monocytes and DC1 antigen presentation and TH1/TH17 immunity upon infection has been described previously [67,69]. Mechanistically, it was observed that an increase in either TNF-α or IFN-γ is sufficient to induce the MLL1 activity which stimulates H3K4 methylation and is required for DC stabilization [67].
Further evidence by Liu et al. supported the potential role of influenza virus NS1 in modulating the JAK-STAT signaling by facilitating the export of DNMT3b from the nucleus to the cytoplasm and its subsequent degradation by K48-linked polyubiquitination. Promoter demethylation leads to the expression of specific JAK-STAT signaling suppressors such as SOCS1, SOCS3, PIAS1 and induces inhibition of interferon signaling in an autocrine or paracrine manner [68].
Acetylation and deacetylation of histones play a significant role in macrophage activation and survival. HDAC mediates the regulation of non-histone proteins involved in mechanisms crucial for cellular functions like DNA repair, replication, P53 signaling, HIF-1α, STAT3 or p65. HDAC macrophages display a pro-inflammatory function by producing pro-inflammatory cytokines like TNF-α, MCP-1, IL-1α, IL-1β and IFN-γ and are a potential target for HDAC inhibitors [69].
The role of HDAC2 in modulating the NF-κB activity plays a significant role in the immune evasion strategy of SARS-CoV-2. Nuclear localization of HDAC2 makes it convenient to inhibit the NF-κB activity, thus altering the monocyte and macrophage function [70]. Overexpression and knockdown study with HDAC5, a Type II HDAC indicates the subsequent activation of TNF-α and MCP-1 in macrophages contributing to the inflammatory response.
Respiratory dysfunctions associated with COVID-19 infection are exacerbated by inflammatory response stimulated by monocyte and macrophages. Monocytes play a crucial role in innate immune response, which migrated to the affected areas and differentiated into macrophage and plays a defensive role by producing pro-inflammatory cytokines like IL-1β, TNF-α, IL-6 and chemokines that facilitate migration [71].
HDAC5 localized in the nucleus and participated in the inflammatory response. However, drugs that could activate the HDAC2 and facilitate nuclear export of HDAC5 could be a novel strategy to control the inflammatory response associated with COVID-19. Current evidence indicates methylxanthine theophylline, macrolide antibiotics, the tricyclic antidepressant nortriptyline, the volatile anaesthetics isoflurane, the phenolic compounds gallic acid and curcumin as well as the plant bioactive molecule andrographolide have the potential to induce the HDAC2 activity by inhibiting PI3K-δ signaling (Figure 2) [72–75].

SARS-CoV-2 protein 3b as an epigenetic modulator
Characterization of viral protein components and their host interacting partners could provide novel insight to understand the molecular basis of immunomodulation strategy. Recent evidence indicates the potential role of SARS-CoV-2 protein 3b an accessory protein component, has been observed to interacts with the host protein machinery like RUNX1b. Given that RUNX1b stimulates transcription of genes involved in definitive hematopoiesis and T cell differentiation cytokines and chemokines including IL-2, IL-3, GM-CSF, MIP-1α, CSFR, etc. [74,75].
In response to SARS-CoV-2 infection protein, 3b stimulates phosphorylation of RUNX1b in an ERK-dependent manner and activates IL-2 promoter [74]. This high degree of molecular linkage of RUNX1 is thought to be mediated by HDAC recruitment and T cell cytotoxic response [76]. The evidence supports the notion that efficient T cell function or exaggerated immune response might be epigenetically controlled.
Histone mimicry as a basis for modulation of gene expression & immune evasion
Coronavirus is enveloped single-stranded positive RNA viruses with a genome size ranging from 26.2 to 31.7 kb. The large capped polyadenylated genome constitutes a set of conserved genes arranged in a particular order: 5′ORF1a-ORF1b-S-ORF3-E-M-N-3′. Among these ORF; ORF1a/b exhibits two-thirds of the genome and produces an mRNA (mRNA1) which encodes different structural and functional protein components.
The structural proteins include: S, E, M and N. The N proteins of CoV-2 consist of three conserved domains: N terminal domain (NTD), C terminal domain (CTD) separated by intrinsically disordered regions (IDR) or RNA binding domains. It was observed that the NTD preferentially binds the 3′ end of viral RNA by electrostatic interaction which stabilizes RNA structure and acts as a chaperone and helps in replication. The CTD plays a vital role in the dimer-dimer association, protein interaction and stress response [77].
The NTD and CTD are separated by IDR or IDP which lack 3D shape in their native conformation. These domains play a crucial role in DNA, RNA and protein binding and enhance the RNA binding activity of NTD and CTD. It was also pointed out that these IDPs play a predominant role in viral adaptation, evasion from the host immune system. Regulation of viral protein synthesis managing the economical use of genetic material via alternative splicing, overlapping genes and antisense transcription [78]. It is surprising to note that the human coronavirus NL63 has 7.3% of these disordered residues [79].
The IDP or IDR are also observed in the mammalian proteome characterized by a predominant hydrophilic amino acid with a low abundance of bulky hydrophobic amino acids. Post-translational modification and sharing of the particular motif of eukaryotic proteins like SLiMs make them ideal for regulating the host defensive strategy.
Short linear motifs commonly known as SLiMs, also the components of eukaryotic linear motifs. SLiMs are observed to be a part of histone proteins and acts as a binding target for readers. As the binding affinity and specificity of SLiMs reside in 2–5 residues, it makes it easy to mimic the host SLiMs. It was observed that the presence of histone H3 like sequence within the C-terminal portion of NS1 of the H3N2 subtype of influenza virus [80].
The role of NS1 protein in suppressing Type I IFN response during infection has been described previously. NS1 protein consists of a sequence of 226-ARSK-229 which resembles the first four amino acids (1-ARTK-4) of histone H3. The tails of influenza may contain PDZ ligand (PL) motifs whereas, SUMOylation sequence in the NS1 tail of H1N1 strain [81]. The PL motif observed in ESEV and EPEV within the NS1 tails of avian-derived influenza strain are significantly associated with pathogenicity and could suppress the antiviral response. The PL motif can attenuate the apoptosis of infected cells and increases the viral load [82].
Furthermore, the histone mimicking ability of NS1 of influenza virus and its modulation of host epigenome has been well explained [80]. The study revealed carboxy-terminus of the H3N2 protein NS1 and tail of histone H3 shares homologs sequence. The NS1 protein interacts with the human PAF1 transcription elongation complex (hPAF1C) and decreases the PAF1-mediated antiviral response in a host. F
urthermore, the binding affinity of histone H3 tail and H3N2 NS1 tail to PAF1 has been explained previously, contributing to RNA elongation co-transcriptional process [80]. Interaction between CHD1 and PAF1 in the regulation of transcriptional elongation has been elucidated previously. WDR5 a core subunit of the human MLL and SET1 histone H3K4 methyltransferase complex and is highly important for global H3K4 methylation and HOX gene activation in human cells. Human CHD1 preferentially binds to H3K4me3 a hallmark of actively transcribed chromatin. Recently, it was reported the CHD1 and WDR5 are a potential target for NS1 protein of influenza A H3N2 subtype possess a histone H3K4 like sequence at its CTD and adapts antiviral response [83].
The bromodomain (BRD) is a conserved structural module of chromatin-associated proteins, and histone acetyltransferases play a dynamic role in regulating chromatin-based gene transcription. BRD specifically binds to the acetylated histones and regulates gene expression [84]. Recent affinity purification-based mass spectrometry results indicate the interaction of E protein of SARS-CoV-2 with BRD-containing proteins BRD2, BRD4 disrupting the activity of BRD histones binding by mimicking histone structure.
The N terminus of histone 2A shares local sequence similarity over an alpha helix about 15 residues some of which are in the transmembrane segment of protein E, which suggests mimicking protein E’s action on histone which disrupts its interaction with BRD2, thus evading from host immune defense. Interaction analysis revealed the affinity of Nsp5 (C145A) with TRMT1 and wild-type Nsp5 with TRMT1 and HDAC2.
Taking both wild-type and catalytic dead constructs (C145A) of Nsp5 of SARS-CoV-2 indicates, wild-type Nsp5 exhibits high confidence interaction with epigenetic regulator HDAC2 predicted a cleavage site between the nuclear localization sequence and HDAC domain and suggested an inhibitory effect of Nsp5 on HDAC2 transport into the nucleus. Furthermore, Nsp5 removes zinc finger and the nuclear localization signal of TRMT1 mediates mitochondrial localization (Figure 2) [83].
Fascinating results obtained by the same study from chemoinformatics data indicate valproic acid and the preclinical candidate apicidin possess HDAC2 inhibitory activity with an affinity of 5 and 120 nM, clinical compounds like ABBV-744 and CPI-0610 on BRD2 and BRD4 with affinities of 2 and 39 nM, respectively.
Coronavirus modulates innate epigenetic signaling
The potential role of the pathogen-related receptor (PRR) and pathogen-associated molecular pattern, TLR, JAK-STAT, NF-κB signaling in orchestration against the viral pathogenesis was previously known. TLR2 mRNA was observed to increase in patients with SARS infection. Previous evidence indicates an increase in NF-κB signaling in response to the TLR2 signaling in monocytes in in vitro conditions [84].
Spike proteins of SARS-CoV are cleaved by cathepsin L, factor Xa and trypsin which cleaves the spike protein into S1 and S2 and permits viral entry into the cytoplasm [85]. In epithelial and fibroblast cells displaying ACE2 receptor induces IL-8 production in response to S protein via the AP-1 pathway [86]. Subsequent evidence indicates crucial roles of specific accessory proteins encoded by MERS-CoV antagonize NF-κB signaling to evade the host defense.
Transport of NF-κB into the nucleus is stimulated by the destruction of IkBs, where NF-κB induces activation of cytokine genes. Upon viral infection activation of TLR and retinoic acids inducible gene like receptors (RLRs) and nucleic acid sensors recognize the pathogen-associated molecular pattern. The role of RIG-1 and MDA-5 via MAVS in priming IkB ubiquitination by recruiting TRAF and TAK1 which induces NF-κB activation [87].
However, previous evidence indicates the viral origin of different proteins and proteases causes inactivation of these adaptor molecules leading to silencing of the NF-κB paving the way for immune evasion [88]. A recent infection experiment of coronavirus with 229E cells indicates that p65 chromatin recruitment is highly crucial in NF-κB and its target gene induction. P65 occupying regions are enhancers elements, promoter-transcription start site regions characterized by increased acetylation of H3 and H4 histones which are stimulated by the activity of transcription factor induced after CoV-2 infection and leads to expression of genes associated with antiviral response [89].
Activation of NF-κB allows the synthesis of the A20 protein required for efficient viral replication [89]. Historically, recruitment of inducible transcription factors to the enhancer region has H3K4me1 and H3K27ac and increases the acetylation of H3K36 and H4K5 in the chromatin structure near the promoter region determines virus-induced host cell response in a nuclear transcription-dependent mechanism. The possible interaction of viral protein components with different DNMTs have been well established [61,90]. The potent role of IL-32 in exerting its antiviral response is confined to its ability by inducing the pro-inflammatory cytokines and differentiation of monocytes into macrophages.
Demethylation in the CREB binding site increases the binding of CREB to the promoter followed by IL-32 transcriptional activation in influenza A virus-infected cells. Influenza virus activates IL-32 expression by activating NF-κB and CREB with site-specific demethylation of CRE in the IL-32 promoter region. Inactivation of DNMT1 and DNMAT3b causes hypomethylation of IL-32 promoter in response to influenza virus infection, indicating a host’s protective mechanism in preventing viral replication [91].
Similar findings were reported by Fang et al. (2012), where the downregulation of DNMT3a and DNMT3b, but not that of DNMT1, involves a COX2 dependent IFN-λ1 production by increase NF-κB signaling. The result of this study indicates increased activity of miRNA (mir29) in A549 cells and PBMC derived from the influenza patients induces PKA-mediated phosphorylation of CREB1 and inhibition of DNMTs activity and contributes to COX2 and PGE2 expression [62].
Mechanistic evidence indicates infection with influenza virus induces an increase in the expression of the host methyltransferase Setdb2, which mediates trimethylation of histone H3 Lys9 (H3K9) at the Cxcl1 promoter and make the host susceptible to superinfection with Streptococcus pneumonia [92]. The epigenetic alterations associated with CoV-2 infection are summarized in Table 1.
| Viral components | Host machinery | Epigenetic change | Response | Ref. |
|---|---|---|---|---|
| Protein 3b | RUNX1b | Recruitment | T cell function cytokine response | [93,94] |
| Covid19 | NF-κB TNF-α MCP-1 | HDAC2, HDAC5 | Inflammation | [95,96] |
| Nsp16 Nsp13 Nsp14 | 2′-O-MT activity 5′-triphosphatase and helicase activity N7MT activity | Methylation and mimic of Cap1 structure | Immune evasion from interferon response by protecting of viral RNA from 5′-3′ exonuclease activity | [97,98] |
| CoV infection | Hsp90 induced mTOR pathway | SMYD2 (Lysine methyltransferase) | Autophagy | [99,100] |
| CoV infection | p65 induced NF-κB activity | H3H4 acetylation | Antiviral response | |
| Cov Infection | BRD2 | Mimics H2A histone | Host immune evasion | [83] |
| Nsp5 | TRMT1 HDAC2 | TRMT1 HDAC2 | Prevents HDAC2 transport to nucleus induces mitochondrial localization of HDAC2 | [83] |
| NL63/ IDP or IDR | Host SLiM protein | Mimics Histone H3 | Immune evasion by surpassing Interferon response | [79,80] |
| MERS-CoV | TNF-α Interferon | H3K27me3 | Antagonizes antigen presentation immune evasion | [70,75] |
| Spike protein | ACE2R | Methylation at CpG site | Viral entry and pathogenesis | [61] |
IDR: Intrinsically disordered region; MERS-CoV: Middle East respiratory syndrome coronavirus.
The epigenetic mechanism controls viral RNA replication
Extracellular vesicles trigger epigenetic reprogramming in the host cell. Virally induced vesicle formation can trigger the multiplicity of infection. CoV-2 Nsp-3, -4 and -6 play a fundamental role in the rearrangement of the host cell membrane and required for the establishment of replication-transcription complexes, called replication organelles, which are nothing but the double-membrane vesicles are characteristic features of all RNA viruses including CoV-2 for making stable infection in the host [101,102].
The marginalization of host cell chromatin, a proliferation of nuclear membrane are the prime events during herpes virus infection. However, such molecular events in CoV-2 vesicle are not reported. Vesicle fusion with other cellular components and chromatin disassembly in a GTPase Ran mediated manner was also described in the previous report [103].
5′ OMT as a molecular target plays a crucial role in evasion from the immune system
The exploitation of host synthetic machinery or encoding own proteins to counteract innate immune response is crucial for establishing a successful infection strategy. Methylation of transcriptome involving different RNAs like tRNA, mRNA, rRNA and other noncoding RNAs is crucial for regulating gene expression. Mechanistically eukaryotic mRNA is capped at 2′-O positions (or Nm where N can be any nucleotide) of the 5′-guanosine cap by methyltransferases (MTases) to distinguish endogenous self-capped RNA from exogenous nonself RNA encoded by pathogen lacking Nm.
This mechanism has been well understood at the molecular level where IFIT1 interferon-induced RNA binding protein mediates this effect by preferentially binding to viral RNAs lacking 2′-O-methylation at their 5′ end and preventing RNA translation. Seminal findings suggest a wide variety of pathogens like flaviviruses, coronavirus, Japanese encephalitis virus, mouse virus, dengue virus, SARS-CoV virus and vaccinia virus adopts such strategy for the propagation of successful replication and evasion from interferon response [104].
IFIT1 has been observed to have a higher affinity for RNA lacking 2′-O methylation, IFIT1 can out-compete eIF4E or eIF4F for binding, remove cap 0 RNA from the actively translating pool [99]. Surprisingly, specific pathogens like coronaviruses encode their own viral 2′-O MTases and mimic the host’s cap1 structure by different distinct mechanisms rendered them a potential target for drug development [105].
ORF1 translated into polyproteins (ppla and ppl1ab) which undergoes co- and post-translational modifications and forms 16 nonstructural proteins nsp 1–16. Bioinformatics study revealed SAM-dependent RNA 2′-O-MT activity of nsp16, where nsp13 function as 5′triphosphatase and helicase activity whereas nsp14 exhibits N7MT activity. This 5′ cap protects the viral RNA from degradation by altering 5′-3′ exonuclease activity and induces its translation by stimulating the preinitiation complex formation.
Although the exact role of SARS-CoV-2 2′-O-MT is unknown, it plays an integral part in a viral replicase-transcriptase complex on its interactions with other viral proteins implicated in the formation of a 3′ terminal protein complex. Recent findings from computational analysis study revealed that dolutegravir and bictegravir are potential drug candidates to target SARS-CoV-2 2′-O-MT activity [100].
Coronavirus uses Hsp90-mediated epigenetic process to hijack the infected cells
Historically, histone modification plays a significant role in the epigenetic silencing of endogenous retroviruses. It was observed that ZFP809, a member of the KRAB-ZFP family, induces the silencing of endogenous retroviruses in a sequence-specific manner via recruitment of heterochromatin-inducing complexes. Epigenetic mark involving Histone 3 Lys9 trimethylation (H3K9me3) in host-associated with tightly inactive repressed chromatin [103].
ZFP809 binding to proline tRNA primer-binding site used by some retroviruses to prime reverse transcription. ZFP809 recruits the KRAB domain binding corepressor KAP1 (TRIM28, TIF1b), which induces silencing via recruitment of histone deacetylases, HP1 and the histone methyltransferase SETDB1 (ESET, KMT1E) [106]. Hsp90 chaperon activity plays a vital role in TRIM28/KAP1-mediated epigenetic silencing of endogenous retroviral elements [97].
Hsp90 in influenza virus by binding to the PB2 subunit enhances the RNA polymerase activity. In the poliovirus, Hsp90 is required for proper folding of the capsid protein. Hsp90 enables viruses to hijack the infected cells through the process of autophagy in targeting the mTOR pathway by inducing the mTOR/p70S6K pathway [98]. It has also been proposed complex interaction between estrogen, Hsp90 and lysine methyltransferases (SMYD2) plays an essential role in autophagy [107]. A similar mechanism might have to go on in SARS-CoV-2-infected patients. Remarkably, a drug repositioning study suggests geldanamycin and its derivatives as the potential candidate to target Hsp90 during COVID-19 infection [95].
reference link: https://www.futuremedicine.com/doi/10.2217/epi-2020-0349
Viruses hijack the host cell ….
Viruses hijack the host cell to replicate their RNA or DNA genomes and create progeny virions. An extreme form of viral parasitism is the integration of a viral genome DNA copy into the host cell DNA (Burns and Boeke, 2012; Feschotte and Gilbert, 2012).
Although diverse classes of RNA viruses create a complementary DNA (cDNA) copy through reverse-transcription of their genomes during their life cycle, integration into the host DNA is a characteristic obligatory step for retroviruses, as well as for endogenous retroelements (Coffin et al., 1997; Burns and Boeke, 2012; Feschotte and Gilbert, 2012).
The machinery that mediates reverse transcription and integration of the retroviral and endogenous retroelement genomes can also use alternative RNA templates, creating genomic cDNA copies of the latter. For example, mammalian apparent long terminal repeat (LTR)-retrotransposons (MaLRs) rely on endogenous retroviruses (ERVs) for their reverse-transcription and integration. Similarly, short interspersed nuclear elements (SINEs), including Alu elements, rely on long interspersed nuclear elements (LINEs) for their reverse transcription and integration (Coffin et al., 1997; Burns and Boeke, 2012; Feschotte and Gilbert, 2012).
The reverse transcriptase and endonuclease activity of LINEs, carried out by the ORF2p protein, can also mediate reverse transcription and integration of unrelated viral and non-viral RNAs (Klenerman et al., 1997; Esnault et al., 2000; Buzdin, 2004). Indeed, the human genome contains DNA copies of distinct RNA and DNA viruses (Blinov et al., 2017), as well as numerous retrogenes and pseudogenes (Baertsch et al., 2008; Richardson et al., 2014; Staszak and Makałowska, 2021), highlighting the possible, albeit infrequent, reverse transcription and integration of non-retroviral RNA into the host genome.
Recent studies reported a high frequency of reverse transcription and integration of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA in infected cells (Zhang et al., 2020; Ying et al., 2021), with implications for diagnostic detection of SARS-CoV-2 nucleic acids by RT-qPCR and for viral antigen persistence.
These findings were partly based on the identification of chimeric reads between viral and human RNA in next-generation RNA-sequencing (RNA-seq) data (Zhang et al., 2020; Ying et al., 2021). Here, we examined the potential source of such chimeric reads and found that they are more likely to be a methodological product, than the result of genuine reverse transcription, integration, and expression.
Discussion
The pandemic caused by SARS-CoV-2 that currently continues to spread globally (Hu et al., 2020), highlighted the need for deeper understanding of its interaction with the human host. The possible genomic integration of SARS-CoV-2 nucleic acids (Zhang et al., 2020; Ying et al., 2021) would have significant implications for host-viral interaction.
The somatic integration of a DNA copy of the RNA virus lymphocytic choriomeningitis virus (LCMV) in the murine host can provide a source of persistent antigen for the immune system (Klenerman et al., 1997). Similarly, persistence of somatically integrated SARS-CoV-2 DNA copies with coding potential could prolong presentation of viral antigens.
However, analyses of intestinal biopsies several months after recovery from COVID-19, indicated the presence of SARS-CoV-2 RNA, as well as presumptive SARS-CoV-2 virions, consistent with on-going replication (Gaebler et al., 2021). Therefore, detection of persistent viral antigen may not necessarily indicate somatic SARS-CoV-2 integration.
Detection of chimeric reads between SARS-CoV-2 RNA and human RNA could also be indicative of somatic SARS-CoV-2 integration. Since detection of such chimeric reads in RNA-seq data would require transcription of the somatic integration, it would likely underestimate the total number of integrations.
The high frequency of expressed somatic SARS-CoV-2 integrations reported (Zhang et al., 2020; Ying et al., 2021) was, therefore, unexpected. However, the majority of chimeric human-SARS-CoV-2 RNA reads may have a different origin. We identified chimeric reads between SARS-CoV-2 RNA and mitochondrial RNA, which were unlikely to have resulted from transcription of SARS-CoV-2 DNA copies integrated into mitochondrial DNA.
If these reads were the result of SARS-CoV-2 integration into mitochondrial DNA, this would require mitochondrial import of viral cDNA and of components of canonical non-homologous end joining (NHEJ) process. While low levels of NHEJ had been reported in mitochondria, no evidence of viral DNA retrotransposition into the mitochondrial genome has yet been reported.
Similarly, we identified chimeric reads between SARS-CoV-2 and RNA transcribed from the adenoviral vector used to overexpress ACE2, in target cells (Blanco-Melo et al., 2020), which would have necessitated integration of SARS-CoV-2 DNA copies in episomal adenoviral DNA. The finding that up to 24% of chimeric reads were formed between SARS-CoV-2 RNA and RNA transcribed from mitochondrial DNA or episomal adenoviral DNA suggested similarly artifactual generation of the remaining reads.
Chimeric reads between nuclear DNA-transcribed RNA and SARS-CoV-2 RNA involved host genes expressed at higher than average level. This correlation may have resulted from more probable detection of the higher expressed, than lower expressed genuine chimeric fragments. Alternatively, it could result from more frequent fortuitous joining, such as during RNA-seq library preparation for example, of SARS-CoV-2 RNA reads with the most abundant host gene RNA reads in the library. In support of the latter possibility, a substantial proportion of chimeric reads displayed complementarity, often over 10 nucleotides, in the joining region.
Moreover, the substantially higher contribution of exonic than intronic or intergenic host sequences to human-SARS-CoV-2 chimeric reads is consistent with formation during RNA-seq library preparation, where exonic sequences are overrepresented relative to intronic or intergenic sequences.
Detection of chimeric reads between SARS-CoV-2 RNA and human RNA is one of several distinct methods previously employed to estimate somatic SARS-CoV-2 integration (Zhang et al., 2020; Ying et al., 2021). Given its dependency on transcription of integrated SARS-CoV-2 cDNA, in addition to the integration step itself, it is likely to be the least sensitive. Direct detection of integrated SARS-CoV-2 cDNA in host genomic DNA, regardless of its expression, was not possible for the datasets used in this study, as whole-genome sequencing data were not available.
Accordingly, the data presented here do not rule out the possibility that SARS-CoV-2 RNA can be reverse-transcribed and integrated in the host DNA. Instead, our study examined specifically the extent to which such integration events can be supported by the detection of chimeric reads between SARS-CoV-2 RNA and human RNA.
At least at the level that can be determined by RNA-seq data analysis, our findings do not indicate frequent genomic integration and subsequent expression of SARS-CoV-2 RNA, and similar conclusions were reached by independent analysis (Yan et al., 2021).
reference link: https://www.frontiersin.org/articles/10.3389/fmicb.2021.676693/full



{kind=link}