









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



L’epigenetica è comunemente definita come lo studio dei cambiamenti fenotipici ereditari senza alterare la sequenza del DNA. Il prefisso greco epi- (ἐπι) in epigenetica implica una funzione “sopra” o “in aggiunta” alla base genetica tradizionale [15].
Negli ultimi due decenni, i regolatori epigenetici sono stati implicati come fattori critici in molti percorsi legati allo sviluppo e alla progressione del cancro e di altre malattie, tra cui la regolazione del ciclo cellulare, l’invasività, le vie di segnalazione, la resistenza alla chemioterapia e l’evasione immunitaria [16,17, 18,19,20,21,22,23,24,25,26,27].
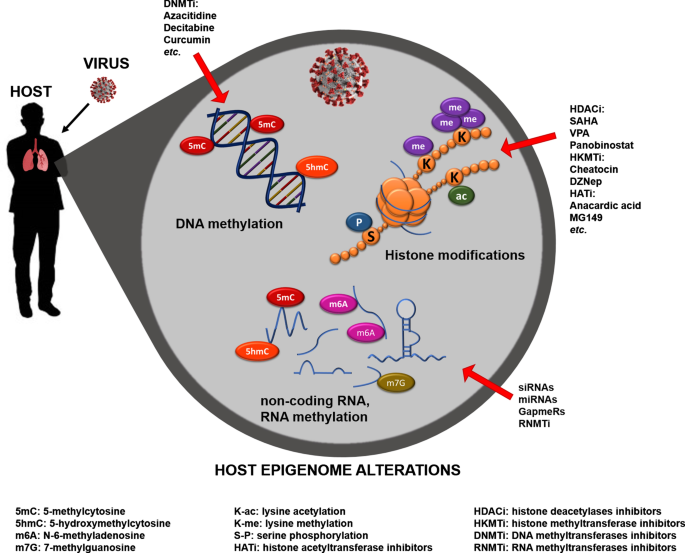
I tre sistemi di base della regolazione epigenetica sono la metilazione del DNA delle regioni di regolazione genica; modificazioni delle proteine istoniche come metilazione, acetilazione, fosforilazione e sumoilazione; e RNA non codificanti [15]. Molte tecniche per l’analisi epigenetica sono già state sviluppate e questo campo è costantemente in fase di innovazione tecnologica [15,28,29].
link di riferimento: https://www.mdpi.com/2227-9059/9/9/1142/htm
L’ Epigenetica
epigenetica è stata descritta come l’area delle scienze della vita che studia il fenotipo ereditabile stabile risultante da cambiamenti negli stati strutturali/di attivazione della cromatina senza alterare la sequenza nucleotidica primaria del DNA [32]. Attualmente, l’epigenomica – lo studio sull’epigenetica del genoma – ci dà la capacità di leggere, localizzare e interpretare funzionalmente il macchinario epigenetico che controlla l’intero genoma a vari livelli [31].
Negli ultimi decenni, un ampio corpus di ricerche ha fornito prove del fatto che l’epigenetica svolge un ruolo importante nell’instaurazione e nella progressione di molte malattie comuni, in particolare quelle legate all’età (malattie legate all’età; ARD) [33]. Inoltre, i modelli di espressione genica determinati dai segni epigenetici molecolari stabiliti durante lo sviluppo influenzano la vulnerabilità a diverse malattie nell’uomo, comprese le infezioni virali [18].
Sorprendentemente, le alterazioni epigenetiche comportano cambiamenti nella struttura della cromatina o nelle proprietà chimiche dell’acido nucleico senza hackerare il codice genetico, a differenza delle mutazioni che alterano direttamente il materiale genetico [32]. Questa caratteristica rende le alterazioni epigenetiche reversibili, flessibili e rapidamente reattive ai cambiamenti ambientali e ad altre esposizioni [34]. Infatti, l’esposizione prolungata a condizioni metaboliche alterate può influenzare epigeneticamente le cellule umane [35].
Diversi meccanismi epigenetici lavorano insieme per regolare l’espressione genica sincronizzando le informazioni metaboliche. A livello della cromatina, la metilazione del DNA e le modificazioni degli istoni portano al rimodellamento della cromatina, e insieme ad altre proteine modificanti (sirtuine, prioni, ecc.) e RNA non codificante (miRNA, sRNA, lncRNA), consentono alla cromatina di accedere alle proteine che regolano il DNA trascrizione e, quindi, sintesi di RNA e proteine [36, 37].
L’epigenoma cellulare riflette lo stato di attivazione genica della cromatina codificando le informazioni su come e dove si trovano e vengono utilizzati gli interruttori di attivazione gene-specifici nel genoma [38]. La cromatina è un complesso di proteine e DNA; il nucleosoma, composto da due copie di quattro istoni core (H3, H4, H2A e H2B), è l’unità fondamentale. Il DNA si avvolge attorno all’ottamero dell’istone, che, grazie alla composizione chimica specifica degli istoni, regola l’accesso al DNA per la trascrizione genica [39].
Il rimodellamento della cromatina controlla molti processi epigenetici attraverso una serie di cambiamenti dinamici nell’organizzazione strutturale del nucleosoma mediante modificazioni reversibili dell’istone e del DNA, che si traducono in diversi livelli di condensazione della cromatina [40]. Tra le centinaia di enzimi coinvolti nella regolazione epigenetica di rilevanza per questo articolo ci sono istone acetiltransferasi (HAT), deacetilasi (HDAC), metiltransferasi (HMT) e chinasi (HK), tutte che agiscono direttamente sui componenti strutturali della cromatina.
Mentre altri, inclusi gli enzimi DNA metiltransferasi (DNMT), le dieci undici proteine di traslocazione (TET) e la timina DNA glicosilasi (TDG), sono coinvolti nel processo attivo di metilazione/demetilazione del DNA. Tutti questi enzimi sono responsabili della creazione di modelli specifici che generano affinità per le proteine associate alla cromatina portando alla loro interazione sinergica o antagonista e determinando le transizioni dinamiche tra stati cromatinici trascrizionalmente attivi o silenti, contribuendo alla plasticità dello sviluppo cellulare e in particolare contesti, portando a esiti patologici [33, 40].
Inoltre, gli ncRNA – la parte trascritta del genoma priva di potenziale codificante per proteine [41] – svolgono un ruolo significativo nella regolazione post-trascrizionale e dell’espressione genica stabilendo o mediando i processi epigenetici, ad esempio silenziando o attivando geni/trascritti attraverso diversi meccanismi di azione. Gli ncRNA possono modulare il comportamento cellulare e sono stati anche implicati come messaggeri intercellulari [42].
Da notare, a seconda della localizzazione subcellulare, gli lncRNA possono mimare i siti di legame del fattore di trascrizione, agendo come esca, o possono legarsi alle giunzioni esone/introne del pre-mRNA e influenzare il processo di splicing [43]. Recentemente, i ricercatori hanno fornito prove sulle modifiche nelle molecole di RNA come la metilazione dell’adenina in posizione 1 (N1-metiladenosina, m1A) e 6 (N6-metiladenosina, m6A) eseguita dalla N6-adenosina metiltransferasi simile 3 (METTL3) [44 ,45,46].
In particolare, la stabilità degli mRNA è alterata quando m6A si verifica alla loro sequenza consenso 5′-AGG (m6) AC-3′, influenzando la loro efficienza di traduzione. Inoltre, come per la 5-metilcitosina (5mC), l’m6A dell’RNA può essere ossidato in N6-idrossimetiladenosina (hm6A) e N6-formiladenosina (f6A) e quindi demetilato, il che potrebbe portare a una diversa interazione RNA-proteina che altera la regolazione genica. A livello di RNA, questi processi sono catalizzati dalla massa grassa e dalla proteina associata all’obesità (FTO) [47].
Degno di nota, RNA e DNA metiltransferasi, come i membri della famiglia del dominio NOP2/Sun e la DNA metiltransferasi di tipo 2 (DNMT2), rispettivamente, possono metilare le ribocitidine in posizione 5 (5 mC) [48, 49]. È interessante notare che gli enzimi TET agiscono in modo simile sulle molecole di DNA e RNA, convertendo l’RNA 5mC in 5-idrossimetilcitosina (5hmC), facilitando la traduzione delle molecole di RNA [50]. Allo stesso modo, i gruppi metilici possono anche essere trasferiti alla posizione 7 della riboguanosina (7-metilguanosina; m7G) [51].
Questa modifica di solito si verifica su mRNA chiusi e ricaptati ed è mediata dalla metiltransferasi canonica mRNA capping (RNMT), che regola la traduzione dell’mRNA in proteine [52]. Da segnalare che, grazie alle moderne tecniche di sequenziamento di nuova generazione, è stato possibile creare mappe dell’epigenoma ad alta risoluzione di cellule sane e malate, consentendo l’analisi simultanea di cambiamenti genetici ed epigenetici, studi di associazione genetica a livello di genoma (GWAS) ed epigenoma -wide associazione studi (EWAS), rispettivamente [53].
Alterazione epigenetica del paesaggio da infezione virale
Tracciare i cambiamenti epigenetici in contesti fisiopatologici potrebbe rappresentare un’interessante fonte di conoscenza per sviluppare nuovi trattamenti che portino, ad esempio, alla regolazione della risposta immunitaria dell’ospite [54]. La maggior parte dei virus appartenenti alla famiglia dei virus corona e influenzali sono solitamente incapaci di hackerare la sequenza genetica dell’ospite, mentre potrebbero alterare l’epigenoma dell’ospite. La ricerca recente si è concentrata su come i virus utilizzano aspetti del meccanismo epigenetico per consentire l’instaurazione, la diffusione e la persistenza dell’infezione [55].
Inoltre, grazie ai recenti progressi nella tecnologia ad alto rendimento, è ora possibile valutare il panorama epigenetico su scala genomica. È stato dimostrato che diverse famiglie di virus antagonizzano il sistema immunitario impiegando una serie di meccanismi epigenetici e, probabilmente, la SARS CoV-2 potrebbe utilizzare la stessa strategia.
Inoltre, diversi rapporti hanno evidenziato come i virus potrebbero interrompere la regolazione della rete epigenetica che ha un impatto sulla risposta immunitaria dell’ospite. Ad esempio, Marazzi et al. hanno mostrato come il virus dell’influenza A H3N2 altamente patogeno inibisca l’inizio della risposta immunitaria innata dell’ospite interferendo con il controllo epigenetico dell’espressione genica [56].
Sfruttando il mimetismo dell’istone, è stato dimostrato che il carbossi-terminale della proteina non strutturale H3N2 NS1 condivide sequenze omologhe con l’amino-terminale della coda dell’istone H3 [56]. In breve, la proteina virale NS1 imita la coda dell’istone dell’istone H3, interagendo con il complesso di trascrizione, che di solito si aggancia al marchio H3K4 per iniziare la trascrizione, interferendo con la funzione del gene antivirale.
Allo stesso modo, sia il virus dell’epatite C (HCV) che gli adenovirus hanno proteine che interferiscono con le funzioni epigenetiche e alterano la funzione immunitaria globale [57, 58]. Nel 2014, il gruppo di Baric ha trovato una chiara associazione di modificazione repressiva dell’istone Trimetilazione dell’istone 3-Lisina 27 (H3K27me3) con geni stimolati dall’interferone (IFN) down-regolati (ISG) a seguito sia del MERS-CoV che dei virus dell’influenza A/influenza/Vietnam/ 1203/2004 (H5N1-VN1203) infezione [59]; di conseguenza, nonostante l’attivazione dei fattori di trascrizione e delle vie di segnalazione, lo stato represso impedisce fisicamente la trascrizione di questi geni.
Più recentemente, Menachery e colleghi hanno anche osservato che la metilazione del DNA svolge un ruolo simile nella perdita di molecole di presentazione dell’antigene a seguito dell’infezione da MERS-CoV e H5N1-VN1203. Allo stesso modo, la metilazione dell’istone è stata trovata coinvolta nell’abbattimento della risposta immunitaria in H5N1, attraverso l’attività della proteina virale NS1 [60]. È importante sottolineare che i loro dati di sequenziamento hanno suggerito che anche altre regioni specifiche del genoma, forse codificanti geni critici coinvolti nell’antagonismo virale, sono anch’esse bersaglio della metilazione. In particolare, la metilazione del DNA era il principale sospetto nella soppressione della produzione di molecole di presentazione dell’antigene in entrambe le malattie [60].
Un altro studio di Schäfer e Baric ha indicato che SARS-CoV e MERS-CoV potrebbero ritardare o compensare il riconoscimento dei patogeni e i livelli di espressione degli ISG codificando proteine uniche che impediscono la risposta di segnalazione immunitaria [18]. Sulla base di come altri virus come il virus dell’immunodeficienza umana 1 (HIV-1) e l’herpes modulano la cromatina, hanno suggerito che anche questi virus più recenti potrebbero agire in modo simile [61, 62].
Gli interferoni sono mediatori essenziali delle azioni antivirali e iniziatori della risposta immunitaria guidata dai patogeni mediante l’inattivazione degli ISG [63, 64] e molti virus potrebbero sviluppare meccanismi antagonisti per superare specifici effettori ISG [65]. Infatti, durante l’infezione, l’IFN e le risposte immunitarie innate sono ampiamente regolate da specifici segni epigenetici, attraverso la manipolazione dell’attività enzimatica epigenetica e la formazione di complessi di rimodellamento della cromatina.
Il meccanismo epigenetico è responsabile non solo dell’innesco e della memoria delle risposte dell’ospite, ma anche di assicurarne il controllo operativo. Fanta et al. ha correlato i livelli di dimetilazione dell’istone 3-lisina 9 (H3K9me2) con l’espressione di IFN in vitro. H3K9me2 è un marchio istonico repressivo che controlla la metilazione del DNA e i processi di formazione dell’eterocromatina. In particolare, il marchio H3K9me2 ostacola l’acetilazione reclutando la famiglia della proteina 1 dell’eterocromatina [66].
Tuttavia, nello studio di cui sopra, Fang et al. hanno dimostrato che i livelli complessivi del marchio H3K9me2 nella regione del promotore dell’interferone di tipo I e l’espressione degli ISG correlano inversamente le cellule dendritiche, definendo questa modifica dell’istone come un importante regolatore della risposta IFN [66, 67].
D’altra parte, il segno della trimetilazione dell’istone 3-Lisina 4 (H3K4me3), comunemente presente nei promotori attivi, è spesso arricchito nelle regioni del promotore dei recettori Toll-like (TLR). Kaikkonen e collaboratori hanno recentemente dimostrato che 60 minuti dopo la stimolazione con lipopolisaccaride (LPS) dei macrofagi e delle cellule dendritiche, l’acetilazione complessiva dell’istone e il legame della polimerasi II (Pol II) a specifici promotori sono aumentati, suggerendo una specifica regolazione epigenetica del sistema immunitario innato. induzione della risposta [68].
Schäfer et al., utilizzando approcci ChIP-PCR, potrebbero determinare l’occupazione differenziale dei segni istonici nei promotori dei geni ISG, dimostrando che le regioni promotrici dei geni ISG contenevano più istoni con segni attivi di monometilazione H3K4 (H3K4me) rispetto al segno repressivo H3K27me3 , favorendo quindi la cromatina aperta e promuovendo la trascrizione attiva e l’espressione di ISG durante l’infezione da H1N1 e SARS-CoV [18].
Altrimenti, nelle cellule infettate da MERS-CoV, Menachery et al. hanno osservato un aumento dei livelli di H3K27me3 e una riduzione dei livelli di H3K4me3 nella regione del promotore di diversi sottoinsiemi di ISG specifici, che non erano sovraregolati. Questi risultati hanno indicato che questi virus avevano sviluppato meccanismi antagonisti per colpire la risposta immunitaria innata dell’IFN [59].
Anche i virus di tipo RNA, come SARS-CoV, mostrano forti associazioni con le modifiche dell’RNA. Ad esempio, è stato scoperto che le modifiche N6-metiladenosina (m6A) e N6,2′-O-dimetiladenosina (m6Am) (m6A/m) svolgono ruoli essenziali nel ciclo di vita virale. In particolare, possono influenzare la struttura e la replicazione del virus, la risposta immunitaria innata dell’ospite e alcune vie di rilevamento innate.
La metilazione dell’RNA m6A è la modifica epitrascrittomica più abbondante degli mRNA eucariotici ed è stata rilevata su trascritti cellulari e virali, regolando numerosi processi biologici, inclusa l’infezione virale [69, 70]. Imam e colleghi hanno suggerito che m6A e il suo macchinario associato regolano il ciclo di vita del virus dell’epatite B (HBV) del DNA, finalizzato attraverso un RNA intermedio, chiamato RNA pregenomico (pgRNA).
Queste osservazioni hanno indicato che m6A regola l’espressione genica dell’HBV e la trascrizione inversa. Infatti, silenziando le metilasi che introducono la modifica m6A all’RNA, hanno osservato un aumento dei livelli di espressione della proteina HBV, mentre la trascrizione inversa del pgRNA sembrava ridotta [71]. Hanno mappato il sito m6A nell’RNA dell’HBV e hanno scoperto che un motivo di consenso m6A conservato situato nella struttura del loop dello stelo epsilon è il sito per la modifica di m6A.
Questo ciclo si trova all’estremità 3′ di tutti gli mRNA dell’HBV e ad entrambe le estremità 5′ e 3′ del pgRNA. Imma et al. ha identificato un sito m6A nell’ansa dello stelo 5′ epsilon di pgRNA mediante analisi mutazionale, rivelando che m6A è necessario per un’efficiente trascrizione inversa di pgRNA. Inoltre, la loro scoperta ha suggerito che la metilazione di m6A dell’ansa dello stelo 3′ epsilon ha provocato la destabilizzazione delle trascrizioni dell’HBV, indicando una doppia funzione regolatoria di m6A per l’RNA dell’HBV.
Considerando che, Tan e colleghi hanno fornito prove che m6A e m6Am dell’RNA messaggero mediano diverse funzioni cellulari esaminando gli epitrascrittomi virali e cellulari m6A/m durante l’infezione latente e litica da herpesvirus associato al sarcoma di Kaposi (KSHV). I trascritti di KSHV sono caratterizzati da un alto livello di modificazioni m6A/m stabilite durante la replicazione latente e litica, conservate durante l’infezione di diversi tipi cellulari [72].
Tan et al. hanno mostrato che durante la replicazione litica, dopo l’atterramento della proteina 2 (YTHDF2) legante l’RNA di YTH N6-metiladenosina, la degradazione dell’RNA di KSHV è compromessa. YTHDF2 si lega ai trascritti virali e media in modo differenziale la loro stabilità. Inoltre, hanno osservato che l’ipometilazione della regione 5′ non tradotta (UTR) indotta da infezione latente da KSHV e l’ipermetilazione del 3′ UTR potrebbero alterare l’epitrascrittoma dell’ospite influenzando i processi di transizione oncogenico ed epiteliale-mesenchimale. Allo stesso tempo, la replicazione litica di KSHV induce una riprogrammazione dinamica dell’epitrascrittoma virale stesso [72].
Infine, il gruppo di Marz ha osservato una consistente firma di metilazione di 5 mC dell’RNA del coronavirus. In particolare, analizzando il contenuto di 5 mC attraverso vari RNA, hanno osservato modelli di metilazione coerenti nelle corrispondenti posizioni genomiche di diversi RNA, suggerendo che la metilazione degli RNA del coronavirus è specifica per la sequenza o controllata da elementi strutturali dell’RNA [73]. La tabella 1 riassume l’implicazione epigenetica nell’infezione virale e i loro esiti funzionali.
Tabella 1 Implicazioni epigenetiche rilevanti nell’infezione virale
| Modificazione epigenetica | Infezione da virus | Obbiettivo | Risultato funzionale |
|---|---|---|---|
| metilazione dell’istone | H3N2 influenza A | H3K4 | Inibizione dell’inizio della risposta immunitaria innata dell’ospite [55] |
| SARS-CoV | H3K4me | Promozione della trascrizione attiva e dell’espressione ISG [16] | |
| H3K4me3 | |||
| H1N1 | H3K4me | Blocco della funzione del gene antivirale [16, 54] | |
| MERS-CoV | H3K27me3 | Down-regulation/inattivazione degli ISG [16, 57, 63] e sviluppo di un meccanismo antagonistico per colpire la risposta immunitaria innata dell’IFN [59] | |
| H3K4me3 | |||
| HSV | – | Down-regulation/inattivazione degli ISG [59, 60] | |
| H5N1-Vn1203 | H3K27me3 | Down-regulation degli ISG [16, 57] | |
| HIV-1 | – | Down-regulation/inattivazione degli ISG [59, 60] | |
| Acetilazione dell’istone | Adenovirus (Ad) E1A | H3K9ac | Interferenza con le funzioni epigenetiche e la funzione immunitaria globale [55] |
| H3K27ac | |||
| metilazione del DNA | SARS-CoV | – | Ritardo/compensazione del riconoscimento del patogeno e modulazione dei livelli di espressione di ISG [16] |
| MERS-CoV | – | Perdita di molecole di presentazione dell’antigene [58] | |
| HSV | – | Ritardo/compensazione del riconoscimento del patogeno e modulazione dei livelli di espressione di ISG [16] | |
| H5N1-Vn1203 | – | Perdita di molecole di presentazione dell’antigene [58] | |
| HIV-1 | – | Ritardo/compensazione del riconoscimento del patogeno e modulazione dei livelli di espressione di ISG [16] | |
| HCV | – | Interferenza con la funzione immunitaria globale [56] | |
| RNA methylation | KSHV | m6A/m6Am | Mediazione della stabilità dei trascritti virali [70] |
| SARS-CoV | 5mC | Modulazione della struttura e della replicazione virale [67, 68] | |
| HBV | m6A | Regolazione dell’espressione genica e trascrizione inversa; destabilizzazione della trascrizione [69] |
Implicazione epigenetica nell’infezione e nella terapia da SARS-CoV-2
Negli ultimi anni, l’epigenetica si è evoluta rapidamente, fornendoci una migliore conoscenza delle funzioni di ereditabilità, dei meccanismi di memoria e della biologia dello sviluppo. Gli studi sull’epigenoma umano stanno diventando sempre più rilevanti in oncologia, immunologia e malattie infettive [74, 75]. In effetti, durante l’ultimo decennio, la ricerca epigenetica ha fornito prove che i virus a DNA e RNA hanno sviluppato funzioni che antagonizzano la macchina regolatrice dell’epigenoma dell’ospite alterando il metabolismo dell’ospite e l’espressione genica, creando un ambiente permissivo per la replicazione e la diffusione del virus [76, 77].
Inoltre, ci sono molte prove che indicano che i cambiamenti legati all’età nell’epigenoma dell’ospite potrebbero compromettere la composizione e la funzione delle cellule immunitarie, influenzando le difese virali, inclusa la risposta immunitaria adattativa [10, 12]. È noto che i coronavirus, come MERS-CoV e SARS-CoV-1, mediano le alterazioni epigenetiche inimicandosi la presentazione dell’antigene dell’ospite o attivando i geni di risposta all’interferone [59, 60].
La valutazione dell’età di metilazione del DNA delle cellule immunitarie e di altri tipi di cellule del sangue prima, durante e dopo l’infezione potrebbe aiutare a spiegare come l’epigenoma invecchiato influisce sulla gravità della malattia e come il virus altera l’epigenoma invecchiato [10]. La vulnerabilità degli anziani alla SARS-CoV-2 potrebbe anche avere a che fare con l’effetto dell’epigenoma sull’ingresso del virus [78]. Questo processo viene avviato sulla superficie cellulare dall’interazione fisica tra il recettore della glicoproteina virale spike, la proteina ACE2 [26] e un corecettore, la dipeptidil peptidasi-4 (DPP4) [30].
Al giorno d’oggi, non esistono ancora farmaci antivirali specifici contro l’infezione da COVID-19 e i vaccini sono ancora in fase di sviluppo. Anche così, sono in fase di studio molti potenziali approcci terapeutici e sono urgentemente necessarie ulteriori ricerche per identificare vaccini efficaci e farmaci sicuri per il trattamento delle infezioni da COVID-19 al fine di sviluppare trattamenti pre e post-esposizione contro l’agente patogeno. Sebbene il primo obiettivo sia la generazione di vaccini basati su SARS-CoV-2 S, con epitopi conservati, in grado di suscitare anticorpi ampiamente neutralizzanti o risposte delle cellule T virus-specifiche, l’identificazione e lo sviluppo di farmaci sicuri ed efficaci per superare SARS-CoV -2 L’immissione e la replica sono essenziali.
Molte strategie per il trattamento del COVID-19 sono state e sono tuttora in fase di studio: diversi farmaci antivirali, tra cui Favipiravir (ClinicalTrials.gov Identifier: NCT04336904), Umifenovir (CTI: NCT04476719) o Lopinavir/Ritonavir (CTI: NCT04386876), da soli o in combinazione con altre sostanze chimiche come l’antimalarico clorochina/idrossiclorochina (es. CTI: NCT04328285); biologici, come plasma convalescente (ad es. CTI: NCT04321421) o cellule staminali mesenchimali (MSC) ed esosomi derivati da MSC (CTI: NCT04276987); Medicinali tradizionali cinesi (es. CTI: NCT04544605) e integrazione con vitamine C e D (CTI: NCT03680274 e NCT04449718; https://www.clinicaltrials.gov/) [79,80,81,82].
La ricerca epigenetica potrebbe aiutare a svolgere questi compiti grazie a una migliore comprensione dei meccanismi coinvolti nella modificazione della cromatina virale nei virus litici e delle interazioni ospite-virus, compresi i fattori genetici che contribuiscono alle risposte protettive o patogene dell’ospite.
Gli studi clinici, gli agenti mirati all’epigenetica approvati dalla FDA e la terapia combinata di farmaci epigenetici e antivirali sono attualmente considerati utili e benefici per la compromissione della replicazione virale e il controllo della risposta immunitaria dell’ospite [83]. Sorprendentemente, le proprietà farmacocinetiche e farmacodinamiche degli antivirali possono anche essere influenzate dalla regolazione epigenetica, evidenziando, ancora una volta, la loro rilevanza nel trattamento dell’infezione da SARS-CoV-2 [84].
Recentemente, El Baba e collaboratori hanno analizzato diversi meccanismi epigenetici coinvolti nelle infezioni da coronavirus, identificando alcuni importanti attori epigenetici che possono essere presi di mira terapeuticamente [83]. Infatti, molte delle proteine non strutturali coinvolte nei processi di trascrizione, replicazione e maturazione virali sono regolate da diverse classi di HDAC, il che implica che gli inibitori HDAC, come Vorinostat o l’acido suberaniloidrossamico (SAHA), combinati con antivirali, potrebbero essere strumenti utili per interferire con questi processi [85, 86].
Da notare che studi precedenti hanno già dimostrato che l’espressione di ACE2 è regolata dalla metilazione del DNA e dalle modifiche dell’istone. In questo contesto, enzimi epigenetici responsabili delle modificazioni sopra menzionate, come DNMT1, istone acetiltransferasi 1 (HAT1), istone deacetilasi 2 (HDAC2) e lisina demetilasi 5B (KDM5B), diventano potenziali bersagli per controllare la risposta immunitaria dell’ospite [87 , 88]. Pertanto, gli inibitori DNMT1, ad esempio l’azacitidina, gli inibitori HAT1, come l’acido anacardico e gli inibitori HDAC2, come l’acido valproico, possono essere riutilizzati contro le infezioni da CoV [79, 82, 89].
Inoltre, sapendo che i virus dipendono dal macchinario epigenetico dell’ospite, i farmaci epigenetici già utilizzati nelle terapie antitumorali potrebbero essere sfruttati per la loro azione antivirale ad ampio spettro e il controllo infiammatorio [83, 90]. In effetti, alcune prove hanno indicato che il principale responsabile delle morti per COVID-19 è la tempesta di citochine, caratterizzata da una sovrapproduzione incontrollata di marcatori solubili dell’infiammazione.
La decitabina o 5-aza-2-deossicitidina (5-azadC), un inibitore del DNMT a base di nucleosidi, è ampiamente utilizzato per inibire la metilazione del DNA nei macrofagi; quindi, sopprimendo l’infiammazione e la risposta all’IFN [83]. Degno di nota, la decitabina è stata recentemente inclusa in uno studio clinico per il trattamento della polmonite-ARDS da COVID-19 (CTI: NCT04482621).
È interessante notare che anche il complesso repressivo polycomb 2 (PRC2), che media la repressione della trascrizione tramite l’arricchimento di H3K27me3 in specifici geni stimolati da IFN, potrebbe essere considerato un bersaglio. Gli inibitori farmacologici della PRC2 sono attualmente in studi clinici avanzati per il trattamento del cancro e potrebbero essere facilmente riutilizzati per trattare i pazienti COVID-19 [91].
Recenti studi mostrano che le cellule immunitarie innate possono possedere una forma di memoria, denominata Trained Immunità (TRIM), un potenziamento a lungo termine della risposta immunitaria innata mantenuta principalmente da cellule natural killer e cellule linfoidi innate del polmone gruppo 2 attraverso meccanismi epigenetici comuni [92, 93]. L’esposizione a uno stimolo iniziale porta queste cellule a una riprogrammazione metabolica, mitocondriale ed epigenetica, che si traduce in un fenotipo di memoria di risposte immunitarie potenziate dopo l’esposizione a uno stimolo eterologo secondario [94].
Geller et al. valutare anche i potenziali effetti del β-glucano sulla disregolazione immunitaria e sulla tempesta di citochine osservate in COVID-19. Nei loro studi, hanno osservato che il TRIM guidato dal β-glucano determina anche alcuni cambiamenti epigenetici e che potrebbe rappresentare un obiettivo utile per il trattamento di COVID-19 [94].
Studi recenti propongono anche vitamine e prodotti naturali, come modificatori epigenetici, per migliorare l’immunità e ridurre la risposta infiammatoria nei pazienti COVID-19 [95,96,97]. Ad esempio, l’uso di vitamina D e quercetina potrebbe essere interessante per migliorare la gravità della SARS-Cov-2 inibendo l’espressione di ACE2 e il suo possibile ruolo nel sopprimere la tempesta di citochine associata alla mortalità nei pazienti COVID-19 [96, 98].
I farmaci a base di RNA sono altri strumenti epigenetici che dovrebbero essere studiati per il trattamento delle infezioni virali [99, 100]. Ad esempio, tra tutti i genomi SARS-CoV che sono stati finora studiati, Baldassarre e collaboratori hanno suggerito che la regione 5′URT e la sua porzione specifica, essenziali per la replicazione e la trascrizione dell’RNA virale, potrebbero essere considerate rilevanti per la progettazione nuove molecole terapeutiche per trattare l’infezione [101].
Nuove strategie che impiegano piccoli RNA interferenti (siRNA), microRNA (miRNA) e oligonucleotidi antisenso dell’acido nucleico bloccati (LNA) o GapmeR, mirando, ad esempio, al 5′URT o alle regioni della molecola Spike, rappresentano potenziali strumenti terapeutici per entrambe le profilassi e la terapia del COVID-19 [101,102,103].
In effetti, la progettazione di oligonucleotidi antisenso, come Miravirsen, in fase di studio per il trattamento dell’HCV, potrebbe essere utilizzata per inibire la replicazione virale mediante lo scavenging dei miRNA coinvolti nel processo [104, 105]. Questi studi suggeriscono che i farmaci a base di RNA potrebbero essere ottimizzati e impiegati per interferire con la replicazione e la trascrizione di SARS-CoV-2. La Figura 2 riassume alcuni dei bersagli epigenetici e degli interventi potenzialmente utili per il trattamento delle infezioni virali da Coronavirus.
Degno di nota, grazie a sofisticati software di bioinformatica, siamo ora in grado di visualizzare e interpretare i dati epigenomici, fornendo approfondite conoscenze cellulari specifiche sulle predisposizioni genetiche ed epigenetiche di un individuo e spiegando come l’ambiente influenzi la funzione dei nostri geni lasciando segni a lungo termine sul genoma. Infatti, la mappatura dell’epigenoma, insieme agli studi EWAS e GWAS, ci fornisce strumenti nella diagnostica di molte malattie umane comuni, indicando che questi studi potrebbero essere impiegati per diagnosi individuali e terapie personalizzate [106, 107].
Pertanto, studiando gli effetti epigenetici del metabolismo dai geni alle vie dei genomi, e grazie alla disponibilità di nuovi dataset epigenomici dettagliati, possiamo esplorare come prevenire, attenuare o invertire terapeuticamente le alterazioni epigenetiche, come progettare e realizzare specifici strumenti farmacologici e quando/dove intervenire. Soprattutto, gli enzimi responsabili delle alterazioni epigenetiche rappresentano un campo entusiasmante per la scoperta di nuovi bersagli farmacologici.
https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-020-00946-x
Approccio epigenetico dell’evasione immunitaria del coronavirus
Le alterazioni epigenetiche di ACE2R determinano l’ingresso di SARS-CoV nella cellula ospite
Mantenere uno stadio di latenza all’interno dell’ospite, imitando il sistema immunitario dell’ospite richiede la manipolazione della cromatina e dell’assemblaggio dell’eterocromatina da parte dei virus. È diventato chiaro che la proteina spike del coronavirus facilita il suo ingresso nelle cellule bersaglio principalmente dall’unità di superficie s1 della proteina S dopo l’innesco della proteina S. L’affinità molecolare tra il recettore ACE2, espresso principalmente sulle cellule epiteliali polmonari di tipo II, è cruciale nell’ingresso virale [47-50]. L’affinità della proteina Spike e del recettore è un determinante cruciale del trofismo tissutale, che svolge un ruolo vitale nell’eziopatogenesi della malattia. Pertanto, è fondamentale comprendere la firma epigenetica del gene ACE2 per controllare la fase iniziale di ingresso e fusione del virus.
Lo studio sull’array di metilazione del DNA a livello di genoma e sulle pipeline di metilazione del chip indica il vario grado di metilazione del DNA del gene ACE2 in diversi sottotipi di tessuto. La metilazione del gene ACE2 più bassa in tre siti CpG (cg04013915, cg08559914, cg03536816) era predominante nelle cellule epiteliali polmonari rispetto ad altri tessuti [51]. Uno studio successivo mostra che le ipometilazioni del gene ACE2 sono per lo più limitate alle femmine rispetto al maschio, suggerendo l’angiotensina
II metabolismo e sua associazione con differenze ormonali o differenze genetiche nel dosaggio cromosomico [52]. Inoltre, l’analisi trascrittomica mostra una mancanza di possibile associazione di ACE2 con razza, età e sesso. Tuttavia, la popolazione di fumatori asiatici mostra un ACE2 più elevato rispetto ai non fumatori, suggerendo un impatto epigenetico sull’attività di ACE2 nel sistema respiratorio [53-55]. Tuttavia, la convalida dell’approccio necessita di dati proteomici.
Precedenti prove hanno anche evidenziato la sovraespressione mediata dall’ipo-metilazione di ACE2 e la sua associazione con l’insorgenza della gravità nei pazienti con lupus eritematoso sistemico, una malattia autoimmune in seguito all’infezione di SARS-CoV-2 con cellule T del sangue periferico [56].
In contrasto con ciò, ulteriori studi mostrano il ruolo potenziale del TNF-α nella regolazione della trascrizione del gene ACE e della complessità patologica nelle cellule endoteliali. I risultati indicano che il TNF-α migliora la metilazione del DNA nel promotore ACE diminuendo l’attività di DNMT, DNMT3a e DNMT3b e TET1 [57]. L’approccio trascrittomico e di biologia del sistema ha rivelato la significativa associazione di una maggiore espressione di ACE2 con RAB1A, HAT1, HDAC2 e KDM5B in pazienti con altre comorbidità come ipertensione, diabete e malattia polmonare ostruttiva cronica [58].
Inoltre, il ruolo dell’istone deacetilasi NAD+ dipendente, SIRT1 nell’induzione dell’attività ACE2 stimolando il promotore ACE2 è stato segnalato durante lo stress energetico indica che SIRT1 potrebbe essere un bersaglio per i farmaci epigenetici nel contesto dell’infezione da COVID-19 [54]. L’analisi dell’arricchimento del percorso ha anche rivelato il potenziale ruolo di KDM5B nella regolazione dell’espressione di diversi geni associati ad ACE2 possibilmente mediante segni epigenetici di acetilazione e metilazione come H3K4me1 e H3K4me3, nonché H3K27ac [56]. Sebbene la letteratura sia scarsa sul pattern epigenetico di COVID-19 ACE2, il meccanismo molecolare che regola l’attività di ACE2 non può essere indebolito per quanto riguarda le attuali pandemie e patogenesi.
Il coronavirus induce la modulazione epigenetica delle cellule immunitarie, altera la presentazione dell’antigene e la risposta all’interferone
È evidente che la generazione di citochine e chemochine antinfiammatorie utili inibisce la replicazione virale e migliora la presentazione dell’antigene [57-59]. La risposta ISG svolge un ruolo di primo piano nel controllo dell’infezione virale per un’efficiente funzione immunitaria. L’IFN di tipo 1 induce una cascata di eventi di segnalazione che provoca la trascrizione di diversi ISG [60,61].
Sebbene le prove siano scarse da SARS-CoV-2 in questa prospettiva, gli studi sull’influenza e su altri virus respiratori a RNA forniscono informazioni significative. È stato scoperto che i virus SARS-CoV-2 e MERS-CoV ritardano significativamente l’espressione dell’ISG. I risultati della trascrittomica e della proteomica nelle cellule Calu3 hanno rivelato diverse firme di espressione ISG virus-specifiche. L’infezione da SARS-CoV-2 delle cellule Calu3 ha rivelato una forte induzione di effettori ISG, ma la risposta è stata significativamente ritardata con il picco di espressione a 48 ore dopo l’infezione. Nel 2012, il MERS-CoV appena emerso ha mostrato un drammatico rilascio di ISG ritardato con effetti visibili a 18 ore dopo l’infezione, per la ridotta espressione di potenziali sottoinsiemi di ISG [62].
Ulteriori prove supportano l’idea che la downregulation degli ISG non sia dovuta a una compromissione della cascata di segnalazione, ma alla modifica dell’istone come la metilazione e l’acetilazione indotte da un agente patogeno. In caso di infezione virale come SARS-CoV-2, l’ospite produce IFN di tipo I e III, che induce il complesso di modulazione dell’istone, che rende la rimozione del segno dell’istone repressivo (H3K27me3) inducendo il segno di attivazione come H3K4me3. Questa conversione della cromatina inattiva in cromatina attiva consente il legame di diversi fattori di trascrizione come STAT1 e IRF7 inducendo così l’espressione di ISG [63,64].
Tuttavia, incorporare modifiche istoniche repressive come H3K27me3 e rimuovere il segno attivo H3K4me3 potrebbe imporre uno stato più condensato della cromatina che impedisce il legame del fattore di trascrizione e quindi riduce l’espressione dell’ISG. Inoltre, l’inibizione o la sottoregolazione di una metilasi H3K79, l’enzima Dot1L associato a una ridotta risposta antivirale e facilita la replicazione virale, suggerisce il suo ruolo cruciale nella risposta antivirale [65].
È stato fornito il ruolo prospettico dell’interleuchina dello studio precedente nella regolazione della firma epigenetica [66]. L’autore ha rivelato che il trattamento con IL-1 induce l’attività di STAT-6; un importante fattore di trascrizione per la segnalazione mediata da IL-4 che si lega al promotore della demetilasi H3K27 Jmjd3. Un livello elevato di Jmjd3 diminuisce la dimetilazione e la trimetilazione di H3K27 (H3K27me2/3), portando all’attivazione trascrizionale del gene marcatore M2. L’associazione significativa di H3K9me2 come soppressore della risposta antivirale inducibile da IFN è stata chiarita da Fang et al. (2012) [67,68].
Tuttavia, l’inattivazione della lisina metiltransferasi G9a, un induttore di H3K9me2, ha portato a un’elevata produzione di IFN, suggerendo che la metiltransferasi potrebbe essere un bersaglio epigenetico terapeutico ideale per sfidare l’invasione virale. È interessante notare che i risultati di Menachery et al. (2018) hanno rivelato una possibile associazione della metilazione globale di H3K27me3 con la sottoregolazione dell’ISG e della metilazione del DNA del gene di presentazione dell’antigene su MERS-CoV e H5N1VN1203 [66].
La possibile associazione di TNF-α e H3K4me3 nell’induzione dell’immunità innata addestrata nei monociti e nella presentazione dell’antigene DC1 e l’immunità TH1/TH17 all’infezione è stata descritta in precedenza [67,69]. Meccanicisticamente, è stato osservato che un aumento di TNF-α o IFN-γ è sufficiente per indurre l’attività MLL1 che stimola la metilazione di H3K4 ed è necessaria per la stabilizzazione delle DC [67].
Ulteriori prove di Liu et al. ha supportato il ruolo potenziale del virus dell’influenza NS1 nella modulazione della segnalazione JAK-STAT facilitando l’esportazione di DNMT3b dal nucleo al citoplasma e la sua successiva degradazione mediante poliubiquitinazione legata a K48. La demetilazione del promotore porta all’espressione di specifici soppressori del segnale JAK-STAT come SOCS1, SOCS3, PIAS1 e induce l’inibizione del segnale dell’interferone in modo autocrino o paracrino [68].
L’acetilazione e la deacetilazione degli istoni svolgono un ruolo significativo nell’attivazione e nella sopravvivenza dei macrofagi. L’HDAC media la regolazione delle proteine non istoniche coinvolte in meccanismi cruciali per le funzioni cellulari come la riparazione del DNA, la replicazione, la segnalazione di P53, HIF-1α, STAT3 o p65. I macrofagi HDAC mostrano una funzione proinfiammatoria producendo citochine proinfiammatorie come TNF-α, MCP-1, IL-1α, IL-1β e IFN-γ e sono un potenziale bersaglio per gli inibitori HDAC [69].
Il ruolo di HDAC2 nella modulazione dell’attività di NF-kB svolge un ruolo significativo nella strategia di evasione immunitaria di SARS-CoV-2. La localizzazione nucleare di HDAC2 rende conveniente inibire l’attività di NF-κB, alterando così la funzione dei monociti e dei macrofagi [70]. Studio di sovraespressione e knockdown con HDAC5, un HDAC di tipo II indica la successiva attivazione di TNF-α e MCP-1 nei macrofagi che contribuiscono alla risposta infiammatoria.
Le disfunzioni respiratorie associate all’infezione da COVID-19 sono esacerbate dalla risposta infiammatoria stimolata da monociti e macrofagi. I monociti svolgono un ruolo cruciale nella risposta immunitaria innata, che migra nelle aree colpite e si differenzia in macrofago e svolge un ruolo difensivo producendo citochine pro-infiammatorie come IL-1β, TNF-α, IL-6 e chemochine che facilitano la migrazione [71 ].
HDAC5 localizzato nel nucleo e partecipato alla risposta infiammatoria. Tuttavia, i farmaci che potrebbero attivare l’HDAC2 e facilitare l’esportazione nucleare dell’HDAC5 potrebbero essere una nuova strategia per controllare la risposta infiammatoria associata a COVID-19. Le prove attuali indicano che la metilxantina teofillina, gli antibiotici macrolidi, l’antidepressivo triciclico nortriptilina, gli anestetici volatili isoflurano, i composti fenolici acido gallico e curcumina, nonché la molecola bioattiva della pianta andrografolide, hanno il potenziale per indurre l’attività HDAC2 inibendo la segnalazione PI3K-δ (Figura 2) [72-75].

SARS-CoV-2 proteina 3b come modulatore epigenetico
La caratterizzazione dei componenti delle proteine virali e dei loro partner che interagiscono con l’ospite potrebbe fornire nuove informazioni per comprendere le basi molecolari della strategia di immunomodulazione. Prove recenti indicano che è stato osservato che il ruolo potenziale della proteina 3b SARS-CoV-2, un componente proteico accessorio, interagisce con il macchinario della proteina ospite come RUNX1b. Dato che RUNX1b stimola la trascrizione di geni coinvolti nell’emopoiesi definitiva e citochine e chemochine di differenziazione delle cellule T tra cui IL-2, IL-3, GM-CSF, MIP-1α, CSFR, ecc. [74,75].
In risposta alla proteina dell’infezione da SARS-CoV-2, 3b stimola la fosforilazione di RUNX1b in modo ERK-dipendente e attiva il promotore di IL-2 [74]. Si pensa che questo alto grado di legame molecolare di RUNX1 sia mediato dal reclutamento di HDAC e dalla risposta citotossica delle cellule T [76]. L’evidenza supporta l’idea che una funzione efficiente delle cellule T o una risposta immunitaria esagerata potrebbero essere controllate epigeneticamente.
Il mimetismo dell’istone come base per la modulazione dell’espressione genica e l’evasione immunitaria
Il coronavirus è un virus a RNA positivo a singolo filamento avvolto con una dimensione del genoma che va da 26,2 a 31,7 kb. Il grande genoma poliadenilato con cappuccio costituisce un insieme di geni conservati disposti in un ordine particolare: 5′ORF1a-ORF1b-S-ORF3-EMN-3′. Tra questi ORF; ORF1a/b esibisce i due terzi del genoma e produce un mRNA (mRNA1) che codifica per diversi componenti proteici strutturali e funzionali.
Le proteine strutturali includono: S, E, M e N. Le proteine N di CoV-2 sono costituite da tre domini conservati: dominio N terminale (NTD), dominio C terminale (CTD) separati da regioni intrinsecamente disordinate (IDR) o legame RNA domini. È stato osservato che il NTD lega preferenzialmente l’estremità 3′ dell’RNA virale mediante interazione elettrostatica che stabilizza la struttura dell’RNA e agisce come chaperone e aiuta nella replicazione. Il CTD svolge un ruolo vitale nell’associazione dimero-dimero, nell’interazione proteica e nella risposta allo stress [77].
Il NTD e il CTD sono separati da IDR o IDP che mancano di forma 3D nella loro conformazione nativa. Questi domini svolgono un ruolo cruciale nel legame a DNA, RNA e proteine e migliorano l’attività di legame all’RNA di NTD e CTD. È stato inoltre sottolineato che questi IDP svolgono un ruolo predominante nell’adattamento virale, nell’evasione dal sistema immunitario dell’ospite. Regolazione della sintesi proteica virale gestendo l’uso economico del materiale genetico tramite splicing alternativo, geni sovrapposti e trascrizione antisenso [78]. È sorprendente notare che il coronavirus umano NL63 ha il 7,3% di questi residui disordinati [79].
L’IDP o IDR si osserva anche nel proteoma dei mammiferi caratterizzato da un amminoacido idrofilo predominante con una bassa abbondanza di amminoacidi idrofobici voluminosi. La modifica post-traduzionale e la condivisione del particolare motivo delle proteine eucariotiche come le SLiM le rendono ideali per regolare la strategia difensiva dell’ospite.
Motivi lineari brevi comunemente noti come SLiM, anche i componenti dei motivi lineari eucariotici. Si osserva che gli SLiM fanno parte delle proteine istoniche e agiscono come un bersaglio vincolante per i lettori. Poiché l’affinità di legame e la specificità degli SLiM risiedono in 2-5 residui, è facile imitare gli SLiM ospiti. È stata osservata la presenza della sequenza simile all’istone H3 all’interno della porzione C-terminale di NS1 del sottotipo H3N2 del virus dell’influenza [80].
Il ruolo della proteina NS1 nella soppressione della risposta dell’IFN di tipo I durante l’infezione è stato descritto in precedenza. La proteina NS1 è costituita da una sequenza di 226-ARSK-229 che assomiglia ai primi quattro amminoacidi (1-ARTK-4) dell’istone H3. Le code dell’influenza possono contenere motivi del ligando PDZ (PL) mentre, la sequenza SUMOylation nella coda NS1 del ceppo H1N1 [81]. Il motivo PL osservato in ESEV ed EPEV all’interno delle code NS1 del ceppo influenzale di derivazione aviaria è significativamente associato alla patogenicità e potrebbe sopprimere la risposta antivirale. Il motivo PL può attenuare l’apoptosi delle cellule infette e aumentare la carica virale [82].
Inoltre, è stata ben spiegata la capacità di mimare gli istoni di NS1 del virus dell’influenza e la sua modulazione dell’epigenoma dell’ospite [80]. Lo studio ha rivelato che il carbossi-terminale della proteina H3N2 NS1 e la coda dell’istone H3 condivide la sequenza degli omologhi. La proteina NS1 interagisce con il complesso di allungamento della trascrizione PAF1 umano (hPAF1C) e diminuisce la risposta antivirale mediata da PAF1 in un ospite. F
inoltre, l’affinità di legame della coda dell’istone H3 e della coda H3N2 NS1 a PAF1 è stata spiegata in precedenza, contribuendo al processo di co-trascrizione di allungamento dell’RNA [80]. L’interazione tra CHD1 e PAF1 nella regolazione dell’allungamento trascrizionale è stata chiarita in precedenza. WDR5 una subunità centrale del complesso MLL umano e dell’istone SET1 H3K4 metiltransferasi ed è molto importante per la metilazione globale di H3K4 e l’attivazione del gene HOX nelle cellule umane. Il CHD1 umano si lega preferenzialmente a H3K4me3, un segno distintivo della cromatina trascritta attivamente. Recentemente, è stato riportato che CHD1 e WDR5 sono un potenziale bersaglio per la proteina NS1 del sottotipo influenzale A H3N2 che possiede una sequenza simile all’istone H3K4 al suo CTD e adatta la risposta antivirale [83].
Il bromodomino (BRD) è un modulo strutturale conservato delle proteine associate alla cromatina e le acetiltransferasi dell’istone svolgono un ruolo dinamico nella regolazione della trascrizione genica basata sulla cromatina. La BRD si lega specificamente agli istoni acetilati e regola l’espressione genica [84]. I recenti risultati della spettrometria di massa basata sulla purificazione dell’affinità indicano l’interazione della proteina E di SARS-CoV-2 con le proteine BRD contenenti BRD2, BRD4 che interrompono l’attività del legame degli istoni BRD imitando la struttura dell’istone.
Il terminale N dell’istone 2A condivide la somiglianza della sequenza locale su un’alfa elica di circa 15 residui, alcuni dei quali si trovano nel segmento transmembrana della proteina E, il che suggerisce di imitare l’azione della proteina E sull’istone che interrompe la sua interazione con BRD2, eludendo così la difesa immunitaria dell’ospite . L’analisi dell’interazione ha rivelato l’affinità di Nsp5 (C145A) con TRMT1 e Nsp5 wild-type con TRMT1 e HDAC2.
Prendendo costrutti morti sia wild-type che catalitici (C145A) di Nsp5 di SARS-CoV-2 indica, Nsp5 wild-type mostra un’interazione ad alta confidenza con il regolatore epigenetico HDAC2 ha predetto un sito di scissione tra la sequenza di localizzazione nucleare e il dominio HDAC e ha suggerito un inibitore effetto di Nsp5 sul trasporto di HDAC2 nel nucleo. Inoltre, Nsp5 rimuove il dito di zinco e il segnale di localizzazione nucleare di TRMT1 media la localizzazione mitocondriale (Figura 2) [83].
Gli affascinanti risultati ottenuti dallo stesso studio dai dati chemioinformatici indicano che l’acido valproico e l’apicidina candidato preclinico possiedono attività inibitoria di HDAC2 con un’affinità di 5 e 120 nM, composti clinici come ABBV-744 e CPI-0610 su BRD2 e BRD4 con affinità di 2 e 39 nM, rispettivamente.
Il coronavirus modula la segnalazione epigenetica innata
Il ruolo potenziale del recettore correlato al patogeno (PRR) e del pattern molecolare associato al patogeno, TLR, JAK-STAT, segnalazione di NF-κB nell’orchestrazione contro la patogenesi virale era precedentemente noto. È stato osservato che l’mRNA di TLR2 aumenta nei pazienti con infezione da SARS. Precedenti prove indicano un aumento della segnalazione NF-κB in risposta alla segnalazione TLR2 nei monociti in condizioni in vitro [84].
Le proteine spike di SARS-CoV vengono scisse dalla catepsina L, dal fattore Xa e dalla tripsina che scinde la proteina spike in S1 e S2 e consente l’ingresso virale nel citoplasma [85]. Nelle cellule epiteliali e fibroblasti che mostrano il recettore ACE2 induce la produzione di IL-8 in risposta alla proteina S attraverso la via AP-1 [86]. Prove successive indicano ruoli cruciali di specifiche proteine accessorie codificate da MERS-CoV che antagonizzano la segnalazione NF-κB per eludere la difesa dell’ospite.
Il trasporto di NF-κB nel nucleo è stimolato dalla distruzione di IkB, dove NF-κB induce l’attivazione dei geni delle citochine. Dopo l’attivazione dell’infezione virale dei recettori TLR e degli acidi retinoici, i geni inducibili come recettori (RLR) e i sensori dell’acido nucleico riconoscono il pattern molecolare associato al patogeno. Il ruolo di RIG-1 e MDA-5 tramite MAVS nell’innesco dell’ubiquitinazione di IkB reclutando TRAF e TAK1 che induce l’attivazione di NF-κB [87].
Tuttavia, prove precedenti indicano che l’origine virale di diverse proteine e proteasi causa l’inattivazione di queste molecole adattatrici che portano al silenziamento dell’NF-κB aprendo la strada all’evasione immunitaria [88]. Un recente esperimento di infezione del coronavirus con cellule 229E indica che il reclutamento della cromatina p65 è estremamente cruciale nell’induzione di NF-kB e del suo gene bersaglio. Le regioni che occupano P65 sono elementi potenziatori, regioni del sito di inizio della trascrizione promotore caratterizzate da un’aumentata acetilazione degli istoni H3 e H4 che sono stimolati dall’attività del fattore di trascrizione indotto dopo l’infezione da CoV-2 e portano all’espressione di geni associati alla risposta antivirale [89] .
L’attivazione di NF-κB consente la sintesi della proteina A20 necessaria per un’efficiente replicazione virale [89]. Storicamente, il reclutamento di fattori di trascrizione inducibili nella regione dell’enhancer ha H3K4me1 e H3K27ac e aumenta l’acetilazione di H3K36 e H4K5 nella struttura della cromatina vicino alla regione del promotore determina la risposta della cellula ospite indotta dal virus in un meccanismo dipendente dalla trascrizione nucleare. La possibile interazione dei componenti della proteina virale con diversi DNMT è stata ben stabilita [61,90]. Il potente ruolo di IL-32 nell’esercitare la sua risposta antivirale è limitato alla sua capacità di indurre le citochine pro-infiammatorie e la differenziazione dei monociti in macrofagi.
La demetilazione nel sito di legame di CREB aumenta il legame di CREB al promotore seguito dall’attivazione trascrizionale di IL-32 nelle cellule infettate dal virus dell’influenza A. Il virus dell’influenza attiva l’espressione di IL-32 attivando NF-κB e CREB con demetilazione sito-specifica di CRE nella regione del promotore di IL-32. L’inattivazione di DNMT1 e DNMAT3b provoca l’ipometilazione del promotore di IL-32 in risposta all’infezione da virus dell’influenza, indicando un meccanismo protettivo dell’ospite nel prevenire la replicazione virale [91].
Risultati simili sono stati riportati da Fang et al. (2012), dove la downregulation di DNMT3a e DNMT3b, ma non quella di DNMT1, comporta una produzione di IFN-λ1 dipendente da COX2 aumentando la segnalazione di NF-κB. Il risultato di questo studio indica una maggiore attività del miRNA (mir29) nelle cellule A549 e PBMC derivato dai pazienti con influenza induce la fosforilazione mediata da PKA di CREB1 e l’inibizione dell’attività di DNMT e contribuisce all’espressione di COX2 e PGE2 [62].
L’evidenza meccanicistica indica che l’infezione con il virus dell’influenza induce un aumento nell’espressione della metiltransferasi ospite Setdb2, che media la trimetilazione dell’istone H3 Lys9 (H3K9) al promotore Cxcl1 e rende l’ospite suscettibile alla superinfezione con polmonite da Streptococcus [92]. Le alterazioni epigenetiche associate all’infezione da CoV-2 sono riassunte nella Tabella 1.
| Componenti virali | Macchinari ospiti | Cambiamento epigenetico | Risposta | Rif. |
|---|---|---|---|---|
| Proteina 3b | RUNX1b | Reclutamento | Funzione delle cellule T risposta alle citochine | [93,94] |
| Covid19 | NF-κB TNF-α MCP-1 | HDAC2, HDAC5 | Infiammazione | [95,96] |
| Nsp16 Nsp13 Nsp14 | Attività 2′-O-MT Attività 5′-trifosfatasi ed elicasi Attività N7MT | Metilazione e mimica della struttura Cap1 | Evasione immunitaria dalla risposta all’interferone proteggendo l’RNA virale dall’attività di esonucleasi 5′-3′ | [97,98] |
| Infezione da CoV | Via mTOR indotta da Hsp90 | SMYD2 (Lisina metiltransferasi) | autofagia | [99,100] |
| Infezione da CoV | p65 ha indotto l’attività di NF-κB | Acetilazione H3H4 | Risposta antivirale | |
| Infezione | BRD2 | Imita l’istone H2A | Evasione immunitaria dell’ospite | [83] |
| Nsp5 | TRMT1 HDAC2 | TRMT1 HDAC2 | Impedisce il trasporto di HDAC2 al nucleo induce la localizzazione mitocondriale di HDAC2 | [83] |
| NL63/IDP o IDR | Proteina SLiM dell’ospite | Imita l’istone H3 | Evasione immunitaria superando la risposta all’interferone | [79,80] |
| MERS-CoV | TNF-α Interferone | H3K27me3 | Antagonizza l’evasione immunitaria della presentazione dell’antigene | [70,75] |
| Proteine Spike | ACE2R | Metilazione al sito CpG | Ingresso virale e patogenesi | [61] |
IDR: regione intrinsecamente disordinata; MERS-CoV: coronavirus della sindrome respiratoria mediorientale.
Il meccanismo epigenetico controlla la replicazione dell’RNA virale
Le vescicole extracellulari innescano la riprogrammazione epigenetica nella cellula ospite. La formazione di vescicole indotta da virus può innescare la molteplicità dell’infezione. CoV-2 Nsp-3, -4 e -6 svolgono un ruolo fondamentale nel riarrangiamento della membrana della cellula ospite e sono necessari per la costituzione di complessi di replicazione-trascrizione, detti organelli di replicazione, che altro non sono che le vescicole a doppia membrana. caratteristiche di tutti i virus a RNA incluso CoV-2 per rendere stabile l’infezione nell’ospite [101,102].
L’emarginazione della cromatina della cellula ospite, una proliferazione della membrana nucleare sono gli eventi principali durante l’infezione da virus dell’herpes. Tuttavia, tali eventi molecolari nella vescicola CoV-2 non sono riportati. La fusione delle vescicole con altri componenti cellulari e il disassemblaggio della cromatina in modo mediato da GTPase Ran sono stati descritti anche nel precedente rapporto [103].
5′ L’OMT come bersaglio molecolare svolge un ruolo cruciale nell’evasione dal sistema immunitario
Lo sfruttamento del macchinario sintetico dell’ospite o la codifica delle proprie proteine per contrastare la risposta immunitaria innata è fondamentale per stabilire una strategia di infezione di successo. La metilazione del trascrittoma che coinvolge diversi RNA come tRNA, mRNA, rRNA e altri RNA non codificanti è cruciale per la regolazione dell’espressione genica. L’mRNA eucariotico meccanicistico è limitato alle posizioni 2′-O (o Nm dove N può essere qualsiasi nucleotide) del cappuccio 5′-guanosina dalle metiltransferasi (MTasi) per distinguere l’RNA auto-incapsulato endogeno dall’RNA esogeno non autonomo codificato dall’agente patogeno privo di Nm.
Questo meccanismo è stato ben compreso a livello molecolare in cui la proteina legante l’RNA indotta dall’interferone IFIT1 media questo effetto legandosi preferenzialmente agli RNA virali privi di 2′-O-metilazione alla loro estremità 5′ e prevenendo la traduzione dell’RNA. I risultati seminali suggeriscono che un’ampia varietà di agenti patogeni come flavivirus, coronavirus, virus dell’encefalite giapponese, virus del topo, virus della dengue, virus SARS-CoV e virus vaccinico adotta tale strategia per la propagazione della replicazione e dell’evasione dalla risposta all’interferone [104].
È stato osservato che IFIT1 ha una maggiore affinità per l’RNA privo di metilazione 2′-O, IFIT1 può competere con eIF4E o eIF4F per il legame, rimuovere l’RNA del cappuccio 0 dal pool di traduzione attiva [99]. Sorprendentemente, agenti patogeni specifici come i coronavirus codificano le proprie 2′-O MTasi virali e imitano la struttura cap1 dell’ospite con diversi meccanismi distinti che li hanno resi un potenziale bersaglio per lo sviluppo di farmaci [105].
ORF1 tradotto in poliproteine (ppla e ppl1ab) che subisce modifiche co- e post-traduzionali e forma 16 proteine non strutturali nsp 1–16. Lo studio di bioinformatica ha rivelato l’attività dell’RNA 2′-O-MT SAM-dipendente di nsp16, dove nsp13 funziona come attività 5′trifosfatasi ed elicasi mentre nsp14 mostra attività N7MT. Questo cappuccio 5′ protegge l’RNA virale dalla degradazione alterando l’attività dell’esonucleasi 5′-3′ e induce la sua traduzione stimolando la formazione del complesso di preiniziazione.
Sebbene il ruolo esatto di SARS-CoV-2 2′-O-MT sia sconosciuto, svolge un ruolo fondamentale in un complesso replicasi-trascrittasi virale sulle sue interazioni con altre proteine virali implicate nella formazione di un complesso proteico terminale 3′. Recenti risultati dello studio di analisi computazionale hanno rivelato che dolutegravir e bictegravir sono potenziali candidati ai farmaci per colpire l’attività SARS-CoV-2 2′-O-MT [100].
Il coronavirus utilizza il processo epigenetico mediato da Hsp90 per dirottare le cellule infette
Storicamente, la modificazione dell’istone gioca un ruolo significativo nel silenziamento epigenetico dei retrovirus endogeni. È stato osservato che ZFP809, un membro della famiglia KRAB-ZFP, induce il silenziamento dei retrovirus endogeni in modo sequenza-specifico tramite il reclutamento di complessi che inducono eterocromatina. Marchio epigenetico che coinvolge la trimetilazione dell’istone 3 Lys9 (H3K9me3) nell’ospite associato a cromatina repressa strettamente inattiva [103].
ZFP809 si lega al sito di legame del primer tRNA della prolina utilizzato da alcuni retrovirus per innescare la trascrizione inversa. ZFP809 recluta il corepressore di legame al dominio KRAB KAP1 (TRIM28, TIF1b), che induce il silenziamento tramite il reclutamento di istone deacetilasi, HP1 e l’istone metiltransferasi SETDB1 (ESET, KMT1E) [106]. L’attività di chaperon di Hsp90 gioca un ruolo vitale nel silenziamento epigenetico mediato da TRIM28/KAP1 di elementi retrovirali endogeni [97].
Hsp90 nel virus dell’influenza legandosi alla subunità PB2 aumenta l’attività della RNA polimerasi. Nel poliovirus, Hsp90 è necessario per il corretto ripiegamento della proteina del capside. Hsp90 consente ai virus di dirottare le cellule infette attraverso il processo di autofagia nel prendere di mira la via mTOR inducendo la via mTOR/p70S6K [98]. È stato anche proposto che la complessa interazione tra estrogeno, Hsp90 e lisina metiltransferasi (SMYD2) svolga un ruolo essenziale nell’autofagia [107]. Un meccanismo simile potrebbe essere necessario nei pazienti con infezione da SARS-CoV-2. Sorprendentemente, uno studio sul riposizionamento dei farmaci suggerisce la geldanamicina e i suoi derivati come potenziale candidato per colpire Hsp90 durante l’infezione da COVID-19 [95].
link di riferimento: https://www.futuremedicine.com/doi/10.2217/epi-2020-0349
I virus dirottano la cellula ospite ….
I virus dirottano la cellula ospite per replicare i loro genomi di RNA o DNA e creare virioni di progenie. Una forma estrema di parassitismo virale è l’integrazione di una copia del DNA del genoma virale nel DNA della cellula ospite (Burns e Boeke, 2012; Feschotte e Gilbert, 2012).
Sebbene diverse classi di virus a RNA creino una copia di DNA complementare (cDNA) attraverso la trascrizione inversa dei loro genomi durante il loro ciclo di vita, l’integrazione nel DNA ospite è un passaggio obbligatorio caratteristico per i retrovirus, così come per i retroelementi endogeni (Coffin et al. , 1997; Burns e Boeke, 2012; Feschotte e Gilbert, 2012).
Il macchinario che media la trascrizione inversa e l’integrazione dei genomi dei retroelementi retrovirali ed endogeni può anche utilizzare modelli di RNA alternativi, creando copie di cDNA genomico di quest’ultimo. Ad esempio, i retrotrasposoni (MaLR) di mammifero apparente lunga ripetizione terminale (LTR) si basano su retrovirus endogeni (ERV) per la loro trascrizione inversa e integrazione. Allo stesso modo, gli elementi nucleari intercalati corti (SINE), inclusi gli elementi Alu, si basano su elementi nucleari intercalati lunghi (LINE) per la loro trascrizione inversa e integrazione (Coffin et al., 1997; Burns e Boeke, 2012; Feschotte e Gilbert, 2012).
L’attività di trascrittasi inversa ed endonucleasi delle LINE, svolta dalla proteina ORF2p, può anche mediare la trascrizione inversa e l’integrazione di RNA virali e non virali non correlati (Klenerman et al., 1997; Esnault et al., 2000; Buzdin, 2004) . Infatti, il genoma umano contiene copie di DNA di distinti virus a RNA e DNA (Blinov et al., 2017), nonché numerosi retrogeni e pseudogeni (Baertsch et al., 2008; Richardson et al., 2014; Staszak e Makałowska, 2021 ), evidenziando la possibile, anche se poco frequente, trascrizione inversa e integrazione dell’RNA non retrovirale nel genoma dell’ospite.
Studi recenti hanno riportato un’alta frequenza di trascrizione inversa e integrazione dell’RNA del coronavirus 2 (SARS-CoV-2) della sindrome respiratoria acuta grave nelle cellule infette (Zhang et al., 2020; Ying et al., 2021), con implicazioni per il rilevamento diagnostico degli acidi nucleici SARS-CoV-2 mediante RT-qPCR e per la persistenza dell’antigene virale.
Questi risultati erano in parte basati sull’identificazione di letture chimeriche tra RNA virale e umano nei dati di sequenziamento dell’RNA di nuova generazione (RNA-seq) (Zhang et al., 2020; Ying et al., 2021). Qui, abbiamo esaminato la potenziale fonte di tali letture chimeriche e abbiamo scoperto che è più probabile che siano un prodotto metodologico, piuttosto che il risultato di una vera trascrizione inversa, integrazione ed espressione.
Discussione
La pandemia causata da SARS-CoV-2 che attualmente continua a diffondersi a livello globale (Hu et al., 2020), ha evidenziato la necessità di una comprensione più profonda della sua interazione con l’ospite umano. La possibile integrazione genomica degli acidi nucleici SARS-CoV-2 (Zhang et al., 2020; Ying et al., 2021) avrebbe implicazioni significative per l’interazione ospite-virale.
L’integrazione somatica di una copia del DNA del virus della coriomeningite linfocitica del virus a RNA (LCMV) nell’ospite murino può fornire una fonte di antigene persistente per il sistema immunitario (Klenerman et al., 1997). Allo stesso modo, la persistenza di copie di DNA SARS-CoV-2 integrate somaticamente con potenziale di codifica potrebbe prolungare la presentazione degli antigeni virali.
Tuttavia, analisi delle biopsie intestinali diversi mesi dopo il recupero da COVID-19, hanno indicato la presenza di RNA SARS-CoV-2, nonché presunti virioni SARS-CoV-2, coerenti con la replicazione in corso (Gaebler et al., 2021). ). Pertanto, il rilevamento dell’antigene virale persistente potrebbe non indicare necessariamente l’integrazione somatica di SARS-CoV-2.
Il rilevamento di letture chimeriche tra RNA SARS-CoV-2 e RNA umano potrebbe anche essere indicativo dell’integrazione somatica di SARS-CoV-2. Poiché il rilevamento di tali letture chimeriche nei dati RNA-seq richiederebbe la trascrizione dell’integrazione somatica, sarebbe probabilmente sottostimato il numero totale di integrazioni.
L’alta frequenza di integrazioni somatiche SARS-CoV-2 segnalate (Zhang et al., 2020; Ying et al., 2021) era, quindi, inaspettata. Tuttavia, la maggior parte delle letture chimeriche di RNA umano-SARS-CoV-2 può avere un’origine diversa. Abbiamo identificato letture chimeriche tra RNA SARS-CoV-2 e RNA mitocondriale, che era improbabile che fossero il risultato della trascrizione di copie di DNA SARS-CoV-2 integrate nel DNA mitocondriale.
Se queste letture fossero il risultato dell’integrazione di SARS-CoV-2 nel DNA mitocondriale, ciò richiederebbe l’importazione mitocondriale del cDNA virale e dei componenti del processo canonico di giunzione finale non omologa (NHEJ). Sebbene nei mitocondri siano stati riportati bassi livelli di NHEJ, non è stata ancora riportata alcuna prova di retrotrasposizione del DNA virale nel genoma mitocondriale.
Allo stesso modo, abbiamo identificato letture chimeriche tra SARS-CoV-2 e RNA trascritto dal vettore adenovirale utilizzato per sovraesprimere ACE2, nelle cellule bersaglio (Blanco-Melo et al., 2020), che avrebbero richiesto l’integrazione del DNA SARS-CoV-2 copie nel DNA adenovirale episomico. La scoperta che fino al 24% delle letture chimeriche si è formata tra l’RNA SARS-CoV-2 e l’RNA trascritto dal DNA mitocondriale o dal DNA adenovirale episomico ha suggerito una generazione artefatta simile delle letture rimanenti.
Le letture chimeriche tra l’RNA trascritto dal DNA nucleare e l’RNA SARS-CoV-2 hanno coinvolto i geni dell’ospite espressi a un livello superiore alla media. Questa correlazione può essere il risultato di un rilevamento più probabile dei frammenti chimerici genuini espressi più elevati rispetto a quelli espressi più bassi. In alternativa, potrebbe derivare da un’unione fortuita più frequente, ad esempio durante la preparazione della libreria RNA-seq, di letture di RNA SARS-CoV-2 con le letture di RNA del gene ospite più abbondanti nella libreria. A sostegno di quest’ultima possibilità, una proporzione sostanziale di letture chimeriche mostrava complementarità, spesso oltre 10 nucleotidi, nella regione di unione.
Inoltre, il contributo sostanzialmente più elevato delle sequenze ospiti esoniche rispetto a quelle introniche o intergeniche alle letture chimeriche di SARS-CoV-2 umano è coerente con la formazione durante la preparazione della libreria RNA-seq, in cui le sequenze esoniche sono sovrarappresentate rispetto alle sequenze introniche o intergeniche.
Il rilevamento di letture chimeriche tra RNA SARS-CoV-2 e RNA umano è uno dei diversi metodi distinti precedentemente impiegati per stimare l’integrazione somatica di SARS-CoV-2 (Zhang et al., 2020; Ying et al., 2021). Data la sua dipendenza dalla trascrizione del cDNA SARS-CoV-2 integrato, oltre alla fase di integrazione stessa, è probabile che sia il meno sensibile. Il rilevamento diretto del cDNA SARS-CoV-2 integrato nel DNA genomico dell’ospite, indipendentemente dalla sua espressione, non è stato possibile per i set di dati utilizzati in questo studio, poiché non erano disponibili dati di sequenziamento dell’intero genoma.
Di conseguenza, i dati qui presentati non escludono la possibilità che l’RNA SARS-CoV-2 possa essere retrotrascritto e integrato nel DNA ospite. Invece, il nostro studio ha esaminato in modo specifico la misura in cui tali eventi di integrazione possono essere supportati dal rilevamento di letture chimeriche tra RNA SARS-CoV-2 e RNA umano.
Almeno al livello che può essere determinato dall’analisi dei dati RNA-seq, i nostri risultati non indicano una frequente integrazione genomica e la successiva espressione dell’RNA SARS-CoV-2 e conclusioni simili sono state raggiunte da analisi indipendenti (Yan et al., 2021 ).
link di riferimento: https://www.frontiersin.org/articles/10.3389/fmicb.2021.676693/full



{kind=link}