









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



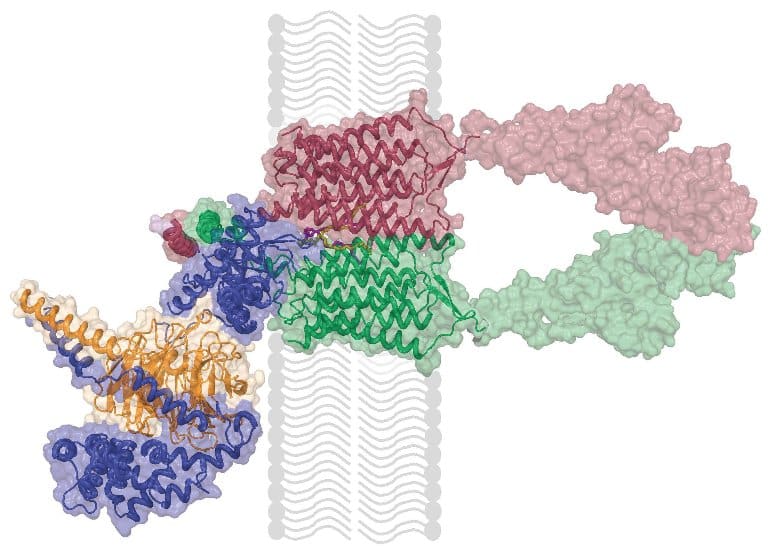
Scientists at Scripps Research, Florida have determined the near-atomic-scale structure of an unusual brain-cell receptor called GPR158, which has been linked to depression and anxiety.
In the study, published Nov. 18 in the journal Science, the researchers used ultracold, single-particle electron microscopy, or cryo-EM, to map, at a resolution of about a third of a billionth of a meter, the atomic structure of GPR158, both on its own and when bound to a group of proteins that mediate its activity.
“We’ve been studying this receptor for more than 10 years, and have done a lot of biology on it, so it’s really gratifying to see for the first time how it’s organized,” says lead author Kirill Martemyanov, Ph.D., Professor and Chair of the Department of Neuroscience at the Scripps Research.
Current treatments work on other known receptors, including monoamine, but don’t always work well for all people and alternative options are needed.
Martemyanov and his team found in a 2018 study that GPR158 is present at unusually high levels in the prefrontal cortex of people diagnosed with major depressive disorder at the time of their death.
They also found that exposing mice to chronic stress increased levels of this receptor in the mouse prefrontal cortex, leading to depression-like behavior—whereas eliminating GPR158 activity in chronically stressed mice made them resistant to depression and the effects of stress. Additionally, the activity of GPR158 receptor has been also linked to prostate cancer.
Historically, GPR158 hasn’t been easy to study. It is called an “orphan receptor” because scientists haven’t yet identified the molecule responsible for turning its signaling function “on” in a manner similar to flipping a switch.
The receptor is also considered unusual because, in the brain, unlike most receptors in its family, it exists in close association with a protein complex called the RGS signaling complex. RGS is short for “regulator of G protein signaling” and it acts as a powerful brake on cellular signaling. However, it has been unclear why GPR158 engages it.
In the new study, solving the receptor’s structure offered many insights into how GPR158 works. First, scientists found that it binds RGS complex in the same way that many receptors typically engage their conventional transducers, leading to the idea that it employs RGS proteins as means of transducing its signal.
“These are fat-related molecules that effectively staple the two halves of the receptor together” Martemyanov explains.
Finally, on the other side of the receptor that faces outside of the cell, an unusual module called the cache domain was revealed.
First author Dipak Patil, Ph.D., a staff scientist in the Martemyanov laboratory, says solving the structure provides many new insights.
The challenge is now to use the information gleaned from the structure to inform the design of small molecule therapeutics to combat depression, Martemyanov adds.
He is now exploring several possible approaches, including disrupting the two-part arrangement, interfering with engagement of RGS complex, or by specifically targeting the cache domain with small, drug-like molecular binders. Regardless of the road taken, availability of structural information should greatly facilitate drug development efforts to treat depression, Martemyanov says.
This study was made possible by the latest technological advances in microscopy, including freezing proteins at ultra-cold temperatures and examining their organization through the lens of powerful microscopes, a technique called cryogenic electron microscopy, or Cryo-EM.
“The microscope uses a beam of electrons instead of light to image protein assemblies. The shorter wavelength of electrons compared to light allowed us to visualize our sample at near-atomic resolution,” says structural biologist Professor Tina Izard, Ph.D.
“The promise of Cryo-EM for achieving significant breakthroughs in solving structures of biomolecules is enormous. Our Institute is firmly committed to expanding Cryo-EM microscopy, which is made possible through the recent acquisition and installation of a new microscope on campus.”
The study was a collaboration including researchers from Columbia University and Appu Singh, Ph.D., a structural biologist at the Indian Institute of Technology in Kanpur.
G protein coupled receptors (GPCRs) are the main mediators of signal transduction in the central nervous system. Therefore, it is not surprising that many GPCRs have long been investigated for their role in the development of anxiety and mood disorders, as well as in the mechanism of action of antidepressant therapies.
Importantly, the endogenous ligands for a large group of GPCRs have not yet been identified and are therefore known as orphan GPCRs (oGPCRs).
Nonetheless, growing evidence from animal studies, together with genome wide association studies (GWAS) and post-mortem transcriptomic analysis in patients, pointed at many oGPCRs as potential pharmacological targets. Among these discoveries, we summarize in this review how emotional behaviors are modulated by the following oGPCRs: ADGRB2 (BAI2), ADGRG1 (GPR56), GPR3, GPR26, GPR37, GPR50, GPR52, GPR61, GPR62, GPR88, GPR135, GPR158, and GPRC5B.
Mood alterations due to pharmacological treatments that modulate serotonergic and noradrenergic systems laid the foundations for the monoamine hypothesis that has led research on mood disorders since the late 1950s [1,2,3]. Dopaminergic alterations have also been associated with major depressive disorder (MDD) symptoms, such as anhedonia [4].
Later, the involvement of the hypothalamic–pituitary–adrenal (HPA) axis was identified, providing a connection between environmental and psychological stressors and the development of affective disorders [5]. Morphological studies in animal models, together with neuroimaging in MDD patients, demonstrated that stress-induced depression is associated with the atrophy of important limbic and cortical brain regions, characterized by the reduction in dendritic arborization, the number of spines, and functional responses [6,7,8,9,10,11,12]; however, increased and sustained amygdala activity, the brain region responsible for the processing of emotions, was also demonstrated, suggesting an altered connectivity among all of these brain areas [7,13,14,15].
Many of these regions express high levels of glucocorticoid receptors and are therefore regulated by the HPA axis through the action of stress hormones on transcriptional programs [16,17]. More recently, evidence of the involvement of further signaling pathways and receptor systems prompted the drug discovery enterprise to move beyond the monoaminergic systems.
Particularly, the observation that the glutamatergic modulator, ketamine, elicits fast-acting antidepressant responses has driven an intensive research effort that culminated with the recent approval of ketamine for treatment-resistant depression, not without raising concerns about its safety [18,19,20,21,22]. The pharmacological treatment of patients diagnosed with bipolar disorder (BPD) consists of mood stabilizing medicines, with lithium remaining the most effective treatment almost seventy years after its serendipitous discovery [23].
The most accredited hypothesis on the mechanism of action of lithium treatments involve the inhibition of inositol monophosphatase and glycogen synthase kinase 3, together with the regulation of calcium signaling, neuroplasticity, neurogenesis, G protein-activated potassium channels, as well as an association with long non-coding RNAs [24,25,26,27,28].
Finally, mood disorders often show comorbidity with anxiety, which is sometimes treated pharmacologically with antidepressants as an alternative to anxiolytic drugs, which indicates the presence of common biological substrates [29]. However, the first choice of pharmacological treatment for anxiety disorders consists in benzodiazepines, in spite of the risk of dependence if taken over a long period of time.
The mechanism of action of benzodiazepines involves the hyperpolarization of neurons, which is achieved by potentiating the action of γ-aminobutyric acid (GABA) on the ligand-gated chloride channels GABAA [30]. With GABA as the main inhibitory neurotransmitter in the central nervous system, benzodiazepine action decreases the firing of action potentials counteracting the neuronal hyperexcitability classically associated with anxiety [30].
In conclusion, despite strong evidence supporting models that include a dysregulation of monoaminergic, glutamatergic, and HPA systems, which are not mutually exclusive, a compelling and unified description of the neurobiology of affective disorders and the mechanisms of action of the medicines used to treat them are still unavailable.
G protein coupled receptors (GPCRs) are 7-transmembrane, metabotropic receptors involved in the regulation of nearly every physiological process. The large GPCR family constitutes the most exploited drug target in the human genome [31,32]. For instance, GPCRs mediate the signaling of neurotransmitters that are targeted by the action of many antidepressants [33,34].
Specifically, treatments with selective serotonin reuptake inhibitors (SSRI) and norepinephrine reuptake inhibitors (NRI), the most common classes of antidepressants, result in an indirect stimulation of serotonin (5-HT) and norepinephrine (NE) GPCRs. Naturally, several clinical and preclinical studies pointed at the modulation of a variety of 5-HT receptors among the mechanisms of action of SSRIs [35].
Similarly, several α-adrenergic receptors for NE have been implicated in the pathophysiology of MDD [36]. However, the pharmacological targeting of single receptors failed to prove an efficient therapy in clinical trials. Over the years, many more GPCRs have been explored as pharmacological targets for the action of novel antidepressants or have been associated with the pathophysiology of MDD and anxiety [33,37,38,39].
Orphan GPCRs (oGPCRs) constitute a subfamily of GPCRs whose endogenous ligands have not yet been identified. oGPCRs represent an important portion of the so-called dark druggable genome, a large group of understudied genes for which biochemical tools and assays are underdeveloped [40].
According to the recommendations of the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR), an oGPCR is considered de-orphanized when reproducibility and in vivo pairing likelihood criteria are met [41,42]. This review focuses on oGPCRs that currently do not meet the NC-IUPHAR criteria and are therefore active subjects of research.
The generation of animal models overexpressing or lacking the expression of specific oGPCRs is a widely used approach to explore their role in mood disorders. Despite the limits in interpreting altered mice behavior as a translation of human affective disorders, numerous tests have been validated pharmacologically and represent a powerful tool to investigate the core symptoms of depression.
Most of the tests for depressive-like behaviors in rodents are based on either learned helplessness, the development of a passive behavior, or behavioral despair, measured as failure to continue in escape-directed efforts after stress. Both the tail suspension test (TST) [43] and the forced swim test (FST) [44] use the measurement of immobility time as an index of a rodent’s effort to escape [45], while the novelty-suppressed feeding test (NSFT) [46,47] relies on the innate fear of rodents for novelty itself.
Anhedonia represents a different aspect of depression that is generally measured in rodents using the sucrose preference test (SPT) [45,48]. On the other side, some of the most widely applied tests for anxiety-related behaviors in rodents are the elevated plus maze (EPM) [49] or elevated zero maze (EZM) [50], the light-dark transition test (LDT) [51], the marble burying test (MBT) [52], and the open field exploration test (OFT) [53], which are all based on the rodents’ innate aversions to brightly illuminated open areas.
These tests have been validated by treatments with anxiolytic drugs. The advantages and drawbacks of each test have been reviewed elsewhere [54,55]. Overall, the behavioral analysis of animal models by applying a combination of the tests remains a fundamental resource to achieve new knowledge on the roles of oGPCRs and their potential to help understand the molecular basis of mood disorders in humans. Unbiased -omics approaches have also been attempted to explore the overall effect of antidepressant treatments, as well as the effect of chronic stress paradigms in animal models [56,57,58].
In addition, comprehensive transcriptomics analyses of post-mortem brain tissues from patients have been carried out to evaluate global changes in gene expression levels [59,60,61]. Finally, genome wide association studies (GWAS) have been addressing the genetic architecture of mood disorders [62,63,64]. This systems biology approach helped revealing the involvement of many oGPCRs in the neurobiology of affective disorders, therefore uncovering alternative potential therapeutic targets.
GPR158
The oGPCR GPR158 shows unique topological features as it lacks the Venus flytrap ligand-binding domain typical of a class C GPCR, while it bears remarkably long extracellular N-terminus and intracellular C-terminus [203]. GPR158 has been proposed as the brain receptor for the bone-derived hormone, osteocalcin [93]. Immunoprecipitation studies indicated a physical interaction and suggested coupling with Gq [93].
Reduced IP1 accumulation in response to osteocalcin treatments of GPR158 KO hippocampal neuronal cultures, as well as altered the electrophysiological effects elicited by osteocalcin treatments in CA3 hippocampal neurons, supported the pairing of GPR158–osteocalcin [93].
However, a number of cellular and molecular adaptations that may be indirectly responsible for the observed altered osteocalcin responses have been reported in the hippocampus and PFC of GPR158 KO mice [92,94,204,205]. Functional in vitro studies are therefore necessary to confirm this ligand–receptor pairing. GPR158 ectodomain has also been shown to interact with several extracellular matrix components of the heparan sulfate proteoglycan family [204,206].
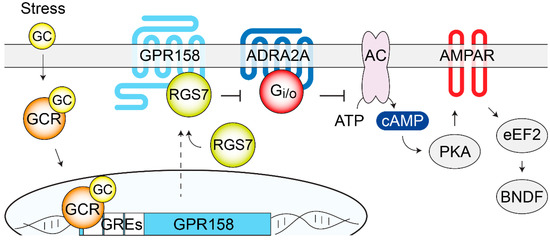
The functional consequences of these interactions on GPR158 signaling are yet to be explored. Proteomics studies identified a G protein-independent signaling mechanism for GPR158, mediated by its interaction with the obligatory heterodimer, Regulator of G protein Signaling 7 (RGS7)/Gβ5 [203], a strong negative modulator of Gi/o signaling.
Specifically, GPR158 has been shown to control proteolytic stability, membrane targeting, and the catalytic activity of RGS7 [207], therefore, forming a unique membrane macromolecular complex that regulates signaling pathways initiated by many other GPCRs in vivo [95]. Finally, quantitative proteomics showed that GPR158 is particularly enriched in the PFC, where it is by far the most expressed oGPCR [92]. In the rest of the brain, GPR158 is also highly expressed in the striatum, amygdala, cerebellum, and hippocampus [93,94,203,204,208].
GPR158 has been recently identified as a key mediator of stress-induced depression, both in mouse models and humans [92,95,205]. A Western blot analysis of post-mortem dorsolateral PFC from diagnosed MDD patients demonstrated significantly higher levels of GPR158 compared to matched controls [92].
Subsequent mouse studies, fostered by the presence of three glucocorticoid response elements in the GPR158 promoter [209], revealed an up-regulation in GPR158 levels by corticosteroid treatments in vivo and in vitro [92]. Similarly, two different protocols of chronic stress, UCMS and PRS, resulted in GPR158 up-regulation in mouse medial PFC (mPFC); an effect that was abolished by the administration of the glucocorticoid receptor blocker RU486 during the stress protocol [92].
Remarkably, the viral overexpression of GPR158 in the mPFC was sufficient to induce a depressive-like state in mice, indicating a primary role in mood regulation played by GPR158, expressed specifically in the mPFC but not in other brain regions [92]. On the contrary, the global ablation of GPR158 expression in mice induced an antidepressant-like behavior [92]. Both male and female adult GPR158 KO mice have been tested in TST and FST revealing a significantly shorter immobility time, which indicates an antidepressant-like phenotype [92].
Notably, such behavior was rescued by the viral overexpression of GPR158 in the mPFC [92]. Furthermore, GPR158 KO mice demonstrated resiliency to chronic stress-induced depression in several behavioral paradigms that also included SPT for anhedonia [92]. Anxiety-like behaviors have also been explored in both male and female GPR158 KO mice using EPM and MBT, revealing an anxiolytic phenotype [92].
Interestingly, the viral up-regulation of GPR158 in the mPFC did not rescue anxiolytic behaviors, indicating that GPR158 expressed in other brain regions must be involved [92]. Opposite results were, however, reported by another group investigating female GPR158 KO mice with EPM, LDT, and OFT [93], while a third research group did not detect any significant difference between the GPR158 KO and wild-type littermates using the OFT [94]. Further research aimed at clarifying the role of GPR158 in anxious behaviors is therefore required. At the cellular level, stress specifically up-regulated GPR158 in the glutamatergic neurons of the mPFC, while the GPR158 levels in GABAergic neurons were not affected [92].
Interestingly, no differences were observed in monoamine levels, or turnover, in the mPFC of the GPR158 KO mice [92]. At the molecular level, it has been demonstrated that GPR158 and RGS7 act as a unique complex in controlling behavioral adaptations, which lead to the modulation of emotional states [92,95]. In fact, male and female RGS7 KO mice showed a phenotype that was identical to what was described for GPR158 KO mice such as antidepressant-like and anxiolytic-like behaviors and resiliency to chronic stress [95].
Notably, the viral down-regulation of RGS7 in the mPFC produced an antidepressant-like phenotype, while RGS7 viral overexpression induced a depressive-like state, as measured by TST and FST [95]. To explore the role played by GPR158 in mediating the behavioral effects observed in RGS7 KO mice, the viral overexpression of RGS7 in the mPFC of GPR158 KO mice was performed, and it was not sufficient to reverse the antidepressant-like phenotype [95]. Moreover, the chronic stress-induced up-regulation of GPR158 in the mPFC augmented the membrane targeting of RGS7 only in the wild-type and not in the GPR158 KO animals [95].
These results point at RGS7 as a key mediator of GPR158 action in controlling depressive-related behaviors. As RGS7 is a known regulator of many GPCRs that have been involved in the action of a variety of antidepressants, pharmacological studies have been performed showing how the inhibition of the GABAB receptors or ADRA2A receptors was sufficient to reverse the antidepressant-like phenotype in GPR158 KO mice [95].
Further evidence from both the GPR158 KO and RGS7 KO studies implicated the intracellular elevation of cAMP levels and changes in the AMPA receptor subunit composition and phosphorylation state, which led to increased AMPA receptor currents [92,95]. The biochemical analysis of signaling proteins involved in the action of antidepressants revealed significant changes in the phosphorylation state of GSK3 and CaMKII in GPR158 KO mice [206].
Moreover, higher BDNF protein levels, that were not paralleled by changes in mRNA levels, were observed in GPR158 KO mPFC, suggesting a regulation of local BDNF translation, a hypothesis supported by a lower phosphorylation of the eukaryotic elongation factor 2 (eEF2) [92]. BDNF was likely responsible for the greater spine density assessed in the mPFC and hippocampal neurons of the GPR158 KO mice [92,204].
Notably, the reported changes in the levels and phosphorylation states of the signaling proteins, together with the alterations in neuronal morphology, are hallmarks of the action of antidepressant treatments and therefore can contribute to the elucidation of the antidepressant-like behaviors of the GPR158 KO mice [210,211], as shown in Figure 4.
Overall, the atypical GPR158–RGS7 complex seems to act as a regulator of traditional GPCRs, which are involved in mediating antidepressant effects, by controlling their volume of G protein signaling. Developing pharmacological means aimed at breaking the complex formation between GPR158 and RGS7 holds promise for novel antidepressant therapies.
reference link : https://www.mdpi.com/2073-4425/11/6/694/htm
Original Research: The findings will appear in Science



{kind=link}