











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


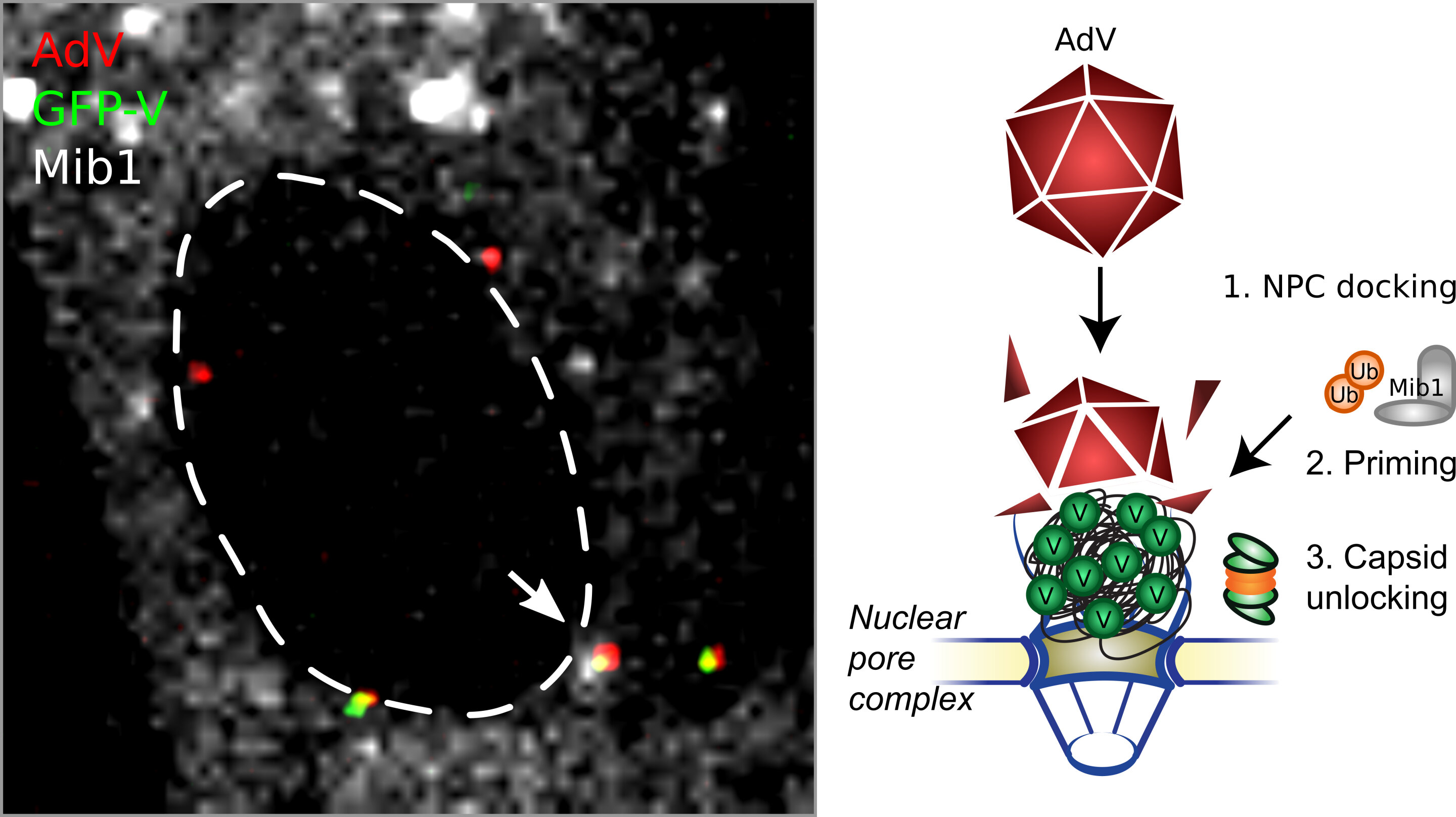
Only then are the genes released into the nucleus, which is necessary for the production of new viruses. This process, discovered by researchers at the University of Zurich, is a key for effective functioning of various COVID-19 vaccines.
Adenoviruses cause respiratory illnesses in humans and have been used as vectors in vaccination for many years, for example against MERS and Ebola virus. Several COVID-19 vaccines are based on replication-defective adenoviruses, including products from AstraZeneca, Johnson & Johnson, CanSino Biologics and Sputnik V.
Viral protein increases stability of virus particle
Researchers are exploiting a key feature of adenoviruses, namely their ability to infect human cells and transfer foreign DNA into the nucleus of these cells. A new study led by Urs Greber, professor at the Department of Molecular Life Sciences at the University of Zurich (UZH) now shows that this process evolved a sophisticated mechanism.
“The viral protein V plays a key role. It connects the DNA with the protein coat surrounding the genome. Protein V increases the stability of the virus particle outside the cell, and also in the cytoplasm of the infected cells,” explains Greber.
The protein coat prevents the cell from recognizing the invading foreign DNA and activating the alarm systems. Once the virus particle reaches the nuclear pore complex—the gateway into the nucleus—the viral DNA is released into the nucleus, where the genetic information is read by the cell’s machinery, resulting in the relevant proteins being produced.
In COVID-19 vaccines, the cells produce the spike protein on the coronavirus’s surface. Presenting the viral protein on the outside of the cell then triggers the immune response in the human body.
Premature release of DNA activates anti-viral alarm systems
The UZH scientists have shown that an adenovirus that is missing the protein V is not only less stable than regular adenoviruses, but also releases its DNA prematurely, before reaching the nuclear pore complex.
“This reduces infection and triggers reactions activating the immune system,” says Greber.
In vector-based vaccines the protective protein coat around the DNA enables the particle to reach the nuclear pore complex, where the viral DNA is released. This “uncoating” is absolutely essential for nuclear import, and success in vector-based vaccination, since the nuclear pore complex prevents large virus particles from invading the nucleus.
Stopping viral infection and improving gene therapy
To demonstrate this key function of Mind bomb1, the researchers used regular adenoviruses to infect human cells that lacked Mind bomb 1, as well as a virus mutant containing protein V, which cannot be modified by this enzyme. In both cases, nuclear import of the viral DNA was defective and the virus particles clustered at the nuclear pore complexes. In other words, the viral infection was stopped.
“Our results inspire the development of anti-viral strategies and enhance procedures to transfer genes into diseased cells for clinical therapy,” says Urs Greber. Notably, adenoviruses are employed in a variety of ways—including use as DNA carriers for gene editing in genetic and metabolic diseases as well as cancer.
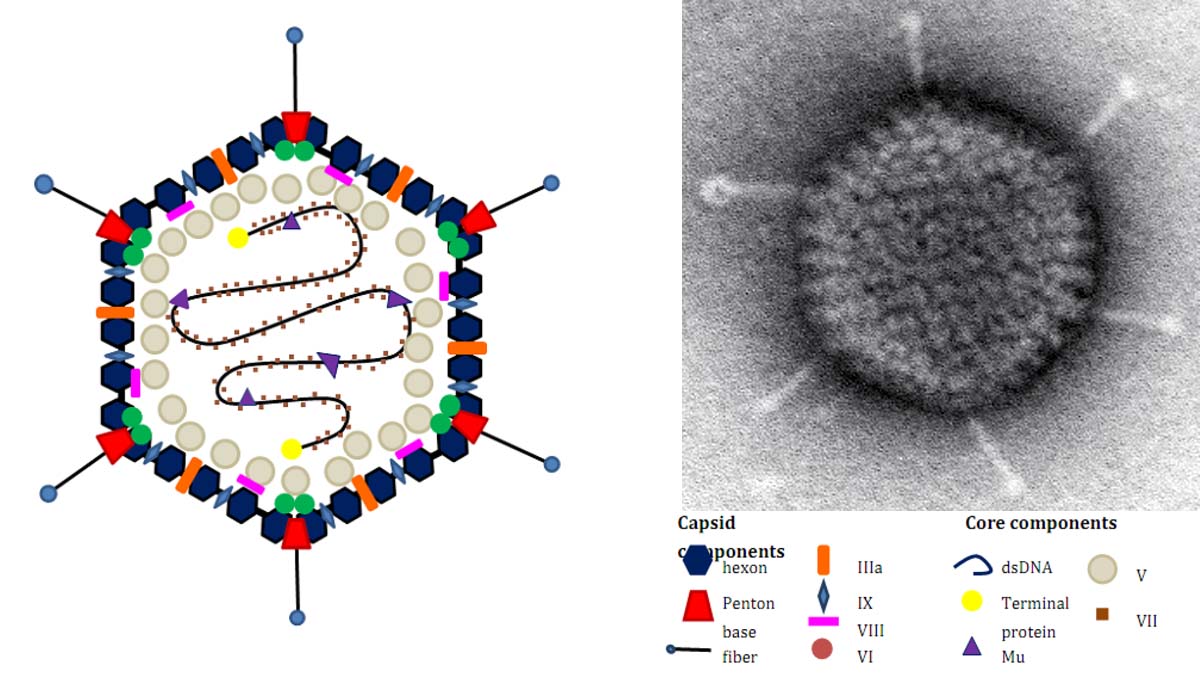
Human adenoviruses are a member of Mastadenovirus, and a non-enveloped, icosahedral virus containing a double-stranded DNA genome (Davison et al., 2003), and contain a few genus-specific genes missing in the other three viral taxons. One of the genes is a minor core protein V (pV) (Davison et al., 2003), and an adenovirus virion contains approximately 157 copies of pV (van Oostrum and Burnett, 1985).
Protein V is speculated to function as a bridge between the core and the capsid proteins (Chatterjee et al., 1985, Matthews and Russell, 1998, Vayda et al., 1983) and non-specifically associated with the double stranded viral genomic DNA (Anderson et al., 1989, Chatterjee et al., 1986, Lehmberg et al., 1999). Initially, pV localizes in the nucleoli during infection, then translocates from the nucleoli to the nucleoplasm as the infection progresses and is incorporated in infectious viral particles (Ugai et al., 2010).
On the other hand, the deletion of the pV gene from the human adenovirus type 5 (Ad5) genome reveals that pV is not essential for adenoviral replication and assembly of infectious viral particles in cancerous cells (Ugai et al., 2007). Interestingly, transient expression of pV induces the redistribution of nucleophosmin 1/NPM1/B23.1 from the nucleoli to the nucleoplasm and the cytoplasm in vitro (Matthews, 2001). Also, adenoviral infection induces NPM1 nucleolar delocalization (Walton et al., 1989).
NPM1 is an abundant nucleolar phosphoprotein (Grisendi et al., 2006), but is expressed at fairly modest levels in human normal cells (Nozawa et al., 1996). In contrast to the expression of NPM1 in human normal cells, NPM1 is isolated as one of the most abundant nuclear matrix proteins in cancer cells (Mattern et al., 1996, Zink et al., 2004), and detected in the nucleoplasm as well as in the nucleoli of tumors (Subong et al., 1999) and overexpressed in various types of tumors (Grisendi et al., 2006).
Thus, the localization and expression of NPM1 are altered in human cancers (Grisendi et al., 2006). On the other hand, NPM1.2 which lacks the 35 amino acids at the C-terminus of NPM1, encodes the RNA binding site, RNase domain, and nucleolar localization signal of NPM1 (Herrera et al., 1995, Hingorani et al., 2000, Wang et al., 1993) expresses in rat tissue and cells at a low level in the cytoplasm and nucleoplasm (Wang et al., 1993).
However, the NPM1.2 expression is not found in human normal cells. Although NPM1.2 expression in human is only detected in HeLa cells which stably express human papillomavirus (HPV) E6 and E7 proteins (Dalenc et al., 2002, Sautkina et al., 2008), the function is largely unclear (Herrera et al., 1995, Savkur and Olson, 1998). NPM1 directly interacts with tumor suppressor p14ARF (ARF) which is an alternative reading frame product of the p16INK4a locus and antagonizes its function in the nucleoli of nonstressed cells (Itahana et al., 2003). In response to cellular and viral stresses, NPM1 is redistributed from the nucleoli to the nucleoplasm and cytoplasm (Hiscox, 2002, Rubbi and Milner, 2003).
There is no evidence that phosphorylation of NPM1 is involved in its translocation during cellular and viral stresses (Kurki et al., 2004). Upon cellular stresses, NPM1 binds to HDM2 which inhibits its E3 ubiquitin ligase activity and protects p53 from HDM2-mediated degradation (Kurki et al., 2004). Also, NPM1 directly interacts with p53 and regulates its protein stability and transcriptional activity (Colombo et al., 2002). Thus, NPM1 functions as a critical regulator of ARF, HDM2 and p53 in the p53 pathway (Grisendi et al., 2006).
Notwithstanding these findings, the precise biological reasons why adenoviral pV redistributes NPM1 in vitro and why NPM1 is redistributed from the nucleoli to the nucleoplasm during infection are not understood. It is also unclear whether pV induces the NPM1 redistribution during infection. Here we demonstrate that pV triggers the relocalization of NPM1 from the nucleoli to the nucleoplasm in primary human endothelial cells by comparative analysis using a pV-deletion mutant, Ad5-dV/TSB, of Ad5. We also provide evidence that adenovirus replication in primary cells requires the relocalization of NPM1 from the nucleoli to the nucleoplasm.
Moreover, we show that NPM1 knockdown inhibits Ad5 replication, demonstrating a direct role of NPM1 in adenoviral replication. While lack of pV causes a great restriction of adenoviral replication in primary cells, Ad5-dV/TSB replicates and induces tumoricidal effect in cancer cells where NPM1 is also abundant in the nucleoplasm. Additionally, transmission electron microscope based analysis demonstrates that lack of pV is linked to a defect in viral assembly in primary cells, but not in cancerous cells. Furthermore, NPM1 is co-purified with and can be directly detected in situ in empty adenovirus particles.
Thus, to our knowledge, NPM1 is the first cellular protein to be identified as being involved in an event of adenoviral virion maturation. These findings imply that at least one significant biological function of pV is in the process of assembly of empty adenovirus particles during virion maturation, and NPM1 is involved in adenoviral biology. Also, our data provide an important new insight in adenoviral assembly and may aid vector design to achieve the selective replication of adenovirus between normal and cancer cells.
Protein V determines the replication property of adenovirus
Lack of E1B-55K mediates a defect in late viral RNA export and late viral protein expression in primary cells as shown by a study on the E1B-55K deletion mutant virus, ONYX-015 (dl1520) (O’Shea et al., 2004). However, dl1520 is an E1b-55K gene deletion mutant derived from an E3-deleted derivative virus called dl309 which is a hybrid of Ad2 and Ad5 sequences in the E1 region (Barker and Berk, 1987).
Thus, dl1520 has a different genetic background compared to the Ad5 genome used in this study. Therefore, we genetically generated dE1B55K in order to determine whether restriction of Ad5-dV/TSB replication depends on reduced expression of E1B-55K. Deletion of the E1B-55K gene showed a dramatic attenuation of late viral protein expression in dE1B55K-infected HPAEC as compared to Ad5- and Ad5-dV/TSB-infected HPAEC cells (Fig. 4A). Despite lack of the E1B-55K expression and a dramatic attenuation of the late viral protein expression in dE1B55K-infected HAPEC cells (Fig. 4A), dE1B55K replication closely paralleled Ad5 replication in HPAEC cells and had a distinct different profile compared to Ad5-dV/TSB replication (Fig. 4B).
Thus, concomitant reduction of E1B55K expression observed in Ad5-dV/TSB-infected cells is not directly responsible for the restriction of Ad5-dV/TSB replication in primary cells. Although we were able to generate dE1B55K-dV/TSB (i.e., deleted for E1B-55K and pV shown in Fig. S1), dE1B55K-dV/TSB virus production was dramatically restricted in A549 cells (see Materials and Methods section). Thus, we were unable to analyze the growth kinetics and protein expression of dE1B55K-dV/TSB.
Also, we did not detect attenuated expression of late viral proteins in Ad5-dV/TSB-infected HPAEC cells as similar to that in dE1B55K-infected HPAEC cells (Fig. 4A). Therefore, the lack of pV does not seem to completely shut off E1B-55K expression in Ad5-dV/TSB-infected HPAEC cells. Based on the kinetics of dE1B55K replication in primary cells (Fig. 4B), we conclude that the attenuation of E1B-55K is not a major determinant of the replication property of Ad5-dV/TSB. Collectively, these data illustrate that pV dictates adenoviral replication in normal cells. Also, these data indicate that the lack of pV, rather than the E1B-55K status determines phenotype of Ad5-dV/TSB replication (Fig. 4).

reference link : https://www.sciencedirect.com/science/article/pii/S0042682212002796?via%3Dihub
More information: Michael Bauer et al, A viral ubiquitination switch attenuates innate immunity and triggers nuclear import of virion DNA and infection, Science Advances (2021). DOI: 10.1126/sciadv.abl7150



{kind=link}