









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


The pleiotropy and complexity of the organ system failures both complicate the care of COVID-19 patients and contribute, to a great extent, to the morbidity and mortality of the pandemic.4
Cardiac manifestations are multifactorial and include hypoxia, hypotension, enhanced inflammatory status, angiotensin-converting enzyme 2 (ACE2) receptor downregulation, endogenous catecholamine adrenergic activation, and direct viral-induced myocardial damage.6, 7
Moreover, patients with underlying cardiovascular disease or comorbidities, including congestive heart failure, hypertension, diabetes, and pulmonary diseases, are more susceptible to infection by SARS-CoV-2, with higher mortality.6, 7
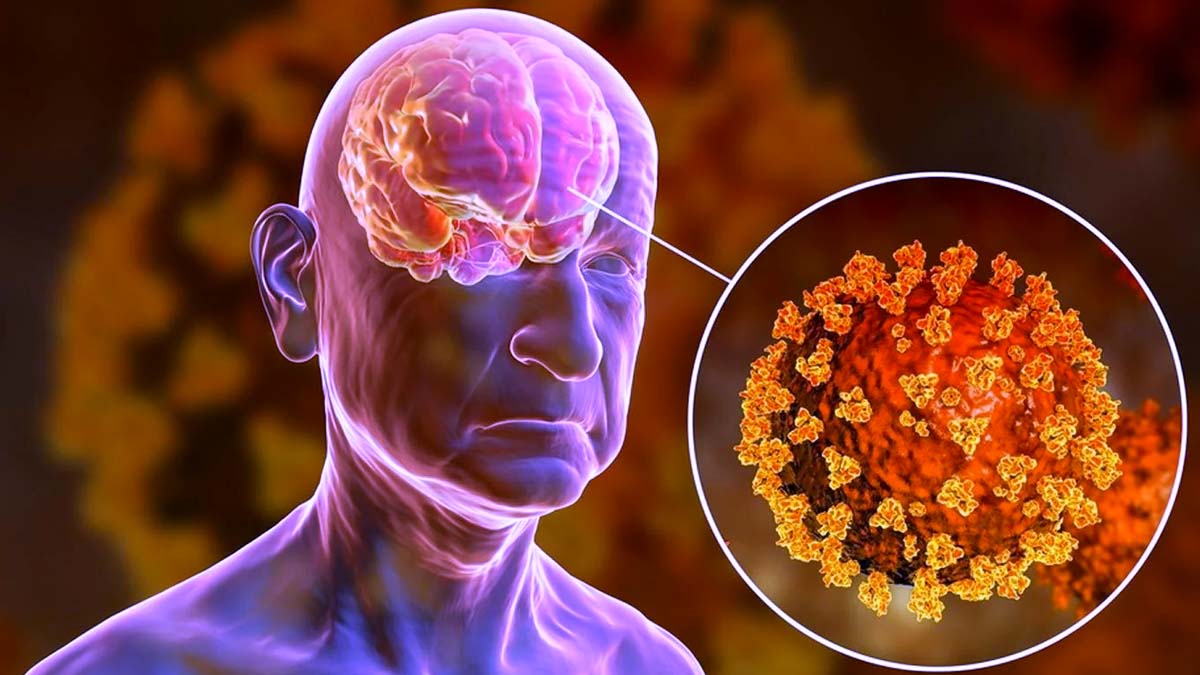
In addition to respiratory and cardiac manifestations, it has been reported that approximately one-third of patients with COVID-19 develop neurological symptoms, including headache, disturbed consciousness, and paresthesias.8 Brain tissue edema, stroke, neuronal degeneration, and neuronal encephalitis have also been reported.2, 8-10
In a recent study, diffuse neural inflammatory markers were found in >80% of COVID-19 patient brains, processes which could contribute to the observed neurological symptoms.11
Furthermore, another pair of frequent symptoms of infection by SARS-CoV-2 are hyposmia and hypogeusia, the loss of the ability to smell and taste, respectively.3
Systemic failure in COVID-19 patients is likely due to SARS-CoV-2 invasion via the ACE2 receptor,9 which is highly expressed in pericytes of human heart8 and epithelial cells of the respiratory tract,12 kidney, intestine, and blood vessels.
ACE2 is also expressed in the brain, especially in the respiratory center and hypothalamus in the brain stem, the thermal center, and cortex,13 which renders these tissues more vulnerable to viral invasion, although it remains uncertain whether SARS-CoV-2 virus directly infects neurons in the brain.14
The primary consequences of SARS-CoV-2 infection are inflammatory responses and oxidative stress in multiple organs and tissues.15-17 Recently it has been shown that the high neutrophil-to-lymphocyte ratio observed in critically ill patients with COVID-19 is associated with excessive levels of reactive oxygen species (ROS) and ROS-induced tissue damage, contributing to COVID-19 disease severity.15
Recent studies have reported an inverse relationship between ACE2 and transforming growth factor-β (TGF-β). In cancer models, decreased levels of ACE2 correlated with increased levels of TGF-β.18 In the context of SARS-CoV-2 infection, downregulation of ACE2 has been observed, leading to increased fibrosis formation, as well as upregulation of TGF-β and other inflammatory pathways.19
Interestingly, reduced angiotensin/ACE2 activity has been associated with tau hyperphosphorylation and increased amyloid beta (Aβ) pathology in animal models of AD.21, 22 The link between reduced ACE2 activity and increased TGF-β and tau signaling in the context of SARS-CoV-2 infection needs further exploration.
Our laboratory has shown that stress-induced ryanodine receptor (RyR)/intracellular calcium release channel post-translational modifications, including oxidation and protein kinase A (PKA) hyperphosphorylation related to activation of the sympathetic nervous system and the resulting hyper-adrenergic state, deplete the channel stabilizing protein (calstabin) from the channel complex, destabilizing the closed state of the channel and causing RyR channels to leak Ca2+ out of the endoplasmic/sarcoplasmic reticulum (ER/SR) in multiple diseases.23-29
Increased TGF-β activity can lead to RyR modification and leaky channels,30 and SR Ca2+ leak can cause mitochondrial Ca2+ overload and dysfunction.29 Increased TGF-β activity31 and mitochondrial dysfunction32 are also associated with SARS-CoV-2 infection.
One consequence of this hyper-adrenergic and oxidative state is the development of tau pathology normally associated with AD. In this article, we investigate potential biochemical pathways linked to tau hyperphosphorylation.
Based on recent evidence that has linked tau pathology to Ca2+ dysregulation associated with leaky RyR channels in the brain,3, 33 we investigated RyR2 biochemistry and function in COVID-19 patient brains.

reference link : https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/alz.12558



{kind=link}