










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


A new study led by researchers from King Abdullah University of Science and Technology (KAUST)-Saudia Arabia has found that mutations in nucleocapsid protein of SARS-CoV-2 affects viral load, host response and disease severity.
The study findings were published in the peer reviewed journal: Nature Communications.
https://www.nature.com/articles/s41467-022-28287-8
The emergence of novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the respiratory coronavirus disease 2019 (COVID-19), resulted in a pandemic that has triggered an unparalleled public health emergency1,2. The global spread of SARS-CoV-2 depended fundamentally on human mobility patterns.
This is highly pertinent to a country like the Kingdom of Saudi Arabia, which as of 22nd February 2021 had a total of 374,691 cases and 6457 deaths 3. The kingdom frequently experiences major population movements, particularly religious mass gatherings.
For instance, during Umrah and Hajj roughly 9.5 million pilgrims visit two Islamic sites in Makkah and Madinah annually 4,5 and the Ministry of Health takes public health measures to keep the pilgrims safe and major outbreaks have been by and large avoided in recent years.
Further, an estimated 5 million Shiite Saudi nationals travel to Iran for pilgrimage, which became an early source of COVID-19 infections in the region 5,6.
Genomic epidemiology of emerging viruses has proven to be a useful tool for outbreak investigation and tracking the pathogen’s progress 8,9. As of October 2021, over four and a half million complete and high coverage genomes are accessible on GISAID 10,11, which aids immensely in tracking the viral sequences globally 12.
Novel SARS-CoV-2 variants are continuously arising and besides providing signals for epidemiological tracking, a subset of the resulting variants will have a functional impact on transmission and infection 13,14,15. It is therefore critical to monitor the genetic viral diversity throughout the pandemic.
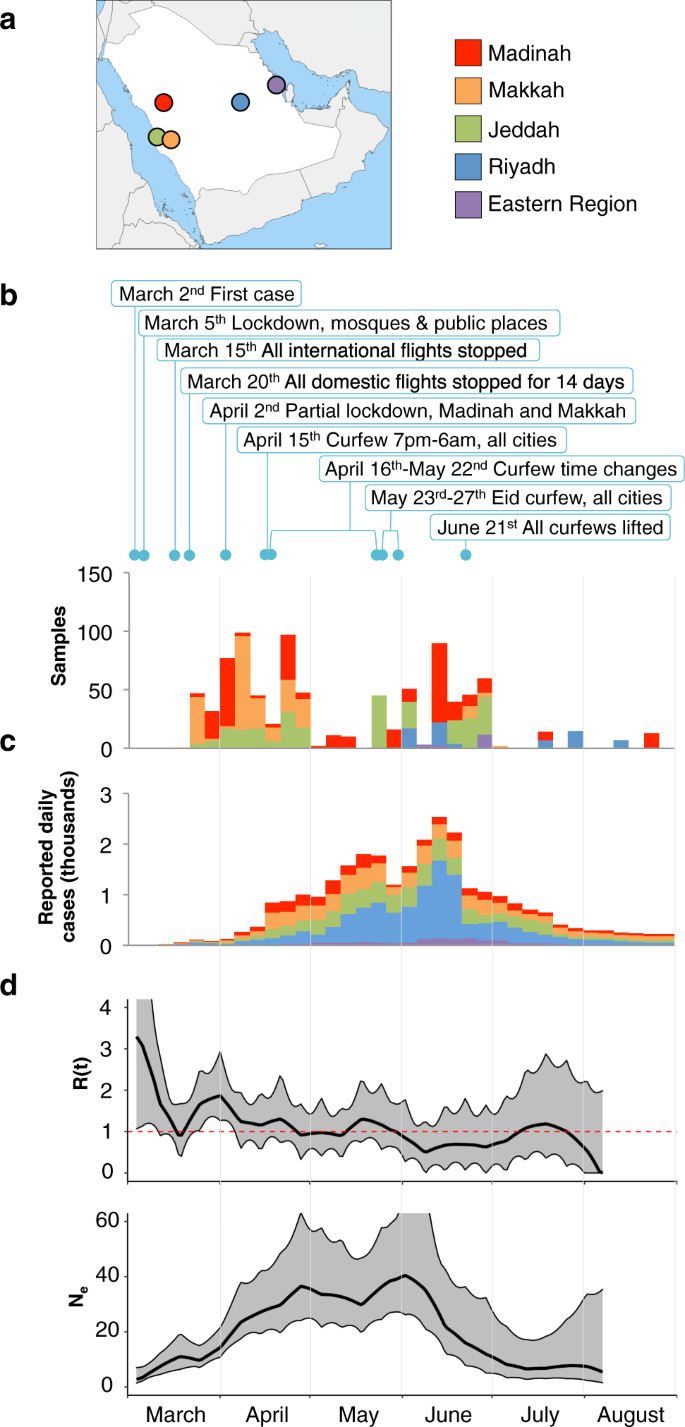
In this study, we sequenced 892 SARS-CoV-2 genomes from nasopharyngeal swab samples of patients from the four main cities, Jeddah, Makkah, Madinah, and Riyadh, as well as a small number of patients from the Eastern region of Saudi Arabia (Fig. 1a, b, Supplementary Data 1, Supplementary Table S1, and Supplementary Table S2).
Samples were mainly collected early in the pandemic and during the first wave of reported cases in Saudi Arabia (Fig. 1c). We analyzed the genomes to investigate the nucleotide changes and multiple mutation events that repres
ent the first 6 months of the locally circulating pandemic lineages of the SARS-CoV-2 in Saudi Arabia and searched for the potential association of polymorphic sites in the genome with available hospital records including severe disease and case fatality rates among the COVID-19 patients.
We performed phylogenetic analysis to visualize the genetic diversity of SARS-CoV-2 and the nature of transmission lineages during March-August, 2020.
We have presented a snapshot of the genomic variation landscapes of the SARS-CoV-2 lineages in our study population and linked a specific set of mutation events in the N gene to viral loads in a diverse population of COVID-19 patients in Saudi Arabia (Supplementary Fig. S1). Finally, we experimentally show the functional impact of these mutations in the N protein on the virus’ interactions with the host.
The R203K/G204R mutations in the N protein affect its interaction with host proteins
According to the SIFT tool37, a substitution at position 204 from G to R in the N protein is predicted to affect functional properties (Fig. 4a). Therefore, we decided to investigate how the two amino acids substitution (R203K and G204R) in the N protein impact its functional interaction with the host that could modulate viral pathogenesis and rewiring of host cell pathways and processes.
HEK-293T cells (three biological replicates) were used for affinity purification followed by mass spectrometry analysis (AP-MS) to identify host proteins associated with the control and mutant N protein (Supplementary Fig. S10). Protein identification was performed using MaxQuant software38.
The compilation of the identified protein groups in mock and N protein (N control and mutant) AP-MS is presented in Supplementary Data 2 (Raw data are available via ProteomeXchange with identifier PXD027168). The majority (62%) of previously reported39 N protein interacting partners overlapped with the identified unadjusted proteins list (Supplementary Fig. S10).
We identified 43 human proteins that displayed significant (adjusted p-value ≤ 0.05, and Log2 fold-change ≥ 1) differential interactions with the mutant and control N protein (Fig. 4d, Supplementary Fig. S10, and see Supplementary Data 3 for both significant and non-significant differentially interacting protein list).
Among these, 42 proteins showed increased interaction and one protein (PRPF19) showed decreased interaction with the N mutant (Fig. 4d and Supplementary Fig. S10). Among the group with increased interaction, we identified many proteins associated with TOR and other signaling pathways (such as AKT1S1 and PIN1), proteins associated with the viral process, viral transcription, and negative regulation of RNA nuclear export (NUP153 and NUP98), and proteins involved in apoptotic and cell death processes (PAWR and ACIN1) (Fig. 4d and Supplementary Fig. S10).
We also identified proteins in the mutant condition that are linked with the immune processes and translation (Fig. 4d and Supplementary Fig. S10). Gene ontology analysis showed that the most enriched biological processes are associated with negative regulation of tRNA and ribosomal subunit export from the nucleus (Fig. 4e).
This finding suggests that the mutant virus may more efficiently inhibit and hijack the host translation to facilitate viral replication and pathogenesis.
Further, many viruses can manipulate the host sumoylation process to enhance viral survival and pathogenesis 40. By pathway enrichment analysis of differentially interacting proteins, we identified pathways associated with sumoylation and antiviral mechanisms (Fig. 4e).
The N mutant (R203K/G204R) induces overexpression of interferon-related genes in transfected host cells
To understand whether the R203K/G204R mutations in the N gene affect host cell transcriptome, we transfected Calu-3 cells (4 biological replicates) with plasmids expressing the full-length N-control and N-mutant protein along with mock-transfection control.
The transcriptome profile of N-mutant and N-control transfected cells displays a distinct pattern from the mock control (Supplementary Fig. S11). We identified 144 and 153 differentially expressed (DE) genes in the N-control and N-mutant transfected cells, respectively, with adjusted p-value < 0.05 and log2 fold-change ≥1 (Supplementary Fig. S11 and Supplementary Data 5).
Among the DE genes, numerous interferon, cytokine, and immune-related genes are up-regulated, some of which are shown in Fig. 5 (for complete list see Supplementary Data 5). We found a robust overexpression of interferon-related genes in the N-mutant compared to N-control transfected cells (Fig. 5a–b) after adjusting for fold-change (Supplementary Fig. S11).
Indeed, strong overexpression of interferon and chemokine-related genes (Supplementary Data 5) were reported in critical COVID-19 patients 46,47. Recent reports further indicate a link between increased expression of interferon-related genes and higher viral load in severe COVID-19 patients 48,49,50. Also, we found overexpression of other genes such as ACE2, STAT147, and TMPRSS1351 (Fig. 5a and Supplementary Data 5) that are elevated in critical COVID-19 disease.
Pathway enrichment analysis (top 15 pathways based on p-value and FDR) of the up-regulated genes (Fig. 5c) shows an overrepresentation of biological processes associated with response to the virus (Fig. 5d and Supplementary Data 6). Similarly, all DE genes were related to substantially enriched pathways, such as interferon-related response, cytokine production, and viral reproductive processes (Supplementary Fig. S11).
The enriched GO terms display an interconnected network highlighting the relationships between up-regulated overlapping genes sets in these pathways (Fig. 5d and Supplementary Fig. S11). Taken together, these results suggest that the R203K/G204R mutations in the N protein may enhance its function in provoking a hyper-expression of interferon-related genes that contribute to the cytokine storm in exacerbating COVID-19 pathogenesis.
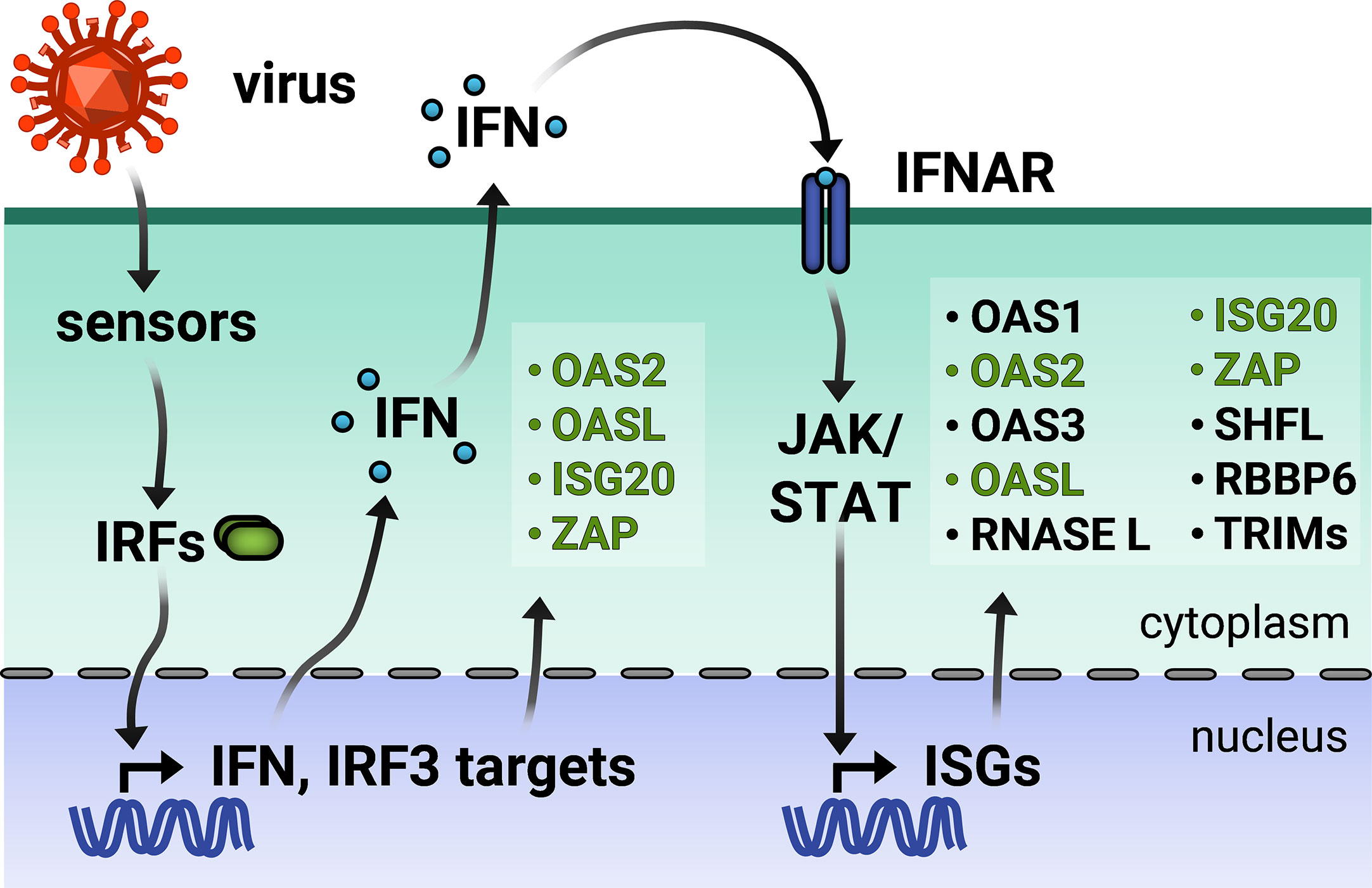
Organisms must constantly defend themselves against viral pathogens. In order to stem viral spread, cells must both signal the presence of viral infection and hinder their replication. One key first line of cellular defense in vertebrates is the type I interferon (IFN) response. Hosts possess sensors which recognize pathogen-associated molecular patterns (PAMPs) of invading viruses such as the viral replicative intermediate double-stranded RNA (dsRNA) and activate transcription factors such as IFN-regulatory factors 3 or 7 (IRF3/7) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). As a result, these transcription factors translocate to the nucleus to activate expression of type I IFN and other proinflammatory cytokines.
The type I IFN receptor is expressed ubiquitously on almost all cell types, allowing for IFN signaling in both infected and neighboring cells that are uninfected. Janus kinase-signal transducer and activator of transcription (JAK-STAT) is the predominant, canonical pathway that regulates ISG transcription. IFN binding to its cell surface receptor, comprised of IFN-α receptor 1 (IFNAR1) and IFN-α receptor 2 (IFNAR2), leads to phosphorylation of pre-associated JAK1. Phosphorylated JAK1 and tyrosine kinase 2 (TYK2) then phosphorylate the IFN receptor, which recruits STAT1/2 to be phosphorylated themselves. Phosphorylated STAT1/2 recruit IRF9 to form the transcription factor complex IFN-stimulated gene factor 3 (ISGF3). ISGF3 translocates to the nucleus where STAT1 is further phosphorylated for full activation. Within the nucleus, ISGF3 binds to IFN-stimulated response elements present in the promoters of IFN-stimulated genes (ISGs), which then effect an antiviral cellular environment [for a comprehensive review on IFN signaling, see (1)].
Interestingly, a growing body of evidence in recent years suggests a plethora of non-canonical mechanisms [for a comprehensive review on canonical and non-canonical regulation of ISG transcription, see (2)]. Non-canonical ISGF3 complexes containing unphosphorylated STAT2, unphosphorylated STAT1 and STAT2, or STAT2 and IRF9 only have been found to mediate expression of specific ISGs (3–6). Other transcription complexes such as STAT5-CrkL (7, 8) or transcription factors such as IRF1 (9) can induce ISG expression. Additionally, cytokines such as TNF-α can moderately induce a subset of ISGs through the NFκB protein complex and further synergize with type I or II IFN to jointly upregulate antiviral ISG expression (10–13). Surprisingly, cellular pathways that have no apparent connection to the innate immune response have been linked to ISG induction. For example, inhibitors of nucleotide synthesis have been shown to effectively upregulate ISG expression in a JAK-STAT-independent manner (14–17). Differences in the extent and timing of ISG upregulation likely depend on the complex interplay between these various mechanisms.
Broadly speaking, an ISG is any gene whose expression is induced by IFN signaling. Advances in RNA-sequencing (RNA-seq) technology have enabled the identification of ISGs across varied cell lines by measuring changes in the transcriptome in response to IFN stimulation. The online database INTERFEROME continues to catalog the results of such gene profiling studies (18). However, ISG expression is more nuanced in reality. A subset of ISGs are direct targets of IRF3/7 and can be induced with or without downstream IFN signaling (Figure 1) (1).
Other ISGs are both basally expressed and IFN-inducible, while still others are cell-type specific (19, 20). Moreover, there are three types of IFN, wherein type I and III are the classic antiviral IFNs. Though type I and III IFNs bind to different receptors, they signal through the same JAK-STAT pathway, thus inducing a shared array of ISGs. Still, type I and III IFN signaling pathways are differentiated by expression kinetics and cell-type specific receptor expression [for a recent review, see (21)]. Tight regulation of ISG expression is necessary because dysregulation of the type I IFN response results in interferonopathies or systemic inflammation deleterious to the organism (22).
In addition to regulating their own expression, ISGs are well known for their inhibition of viral replication. They employ diverse mechanisms to block virtually every step of viral replication, though ISGs have been shown to target different viral life cycle stages for different viruses [for recent reviews on a broad range of antiviral ISGs, see (20, 23)]. For example, the IFITM family blocks viral entry of diverse viruses (24) while the Mx GTPases recognize diverse nucleocapsids and block their nuclear import (25). TRIM5α disrupts retrovirus uncoating and targets several viral proteins for proteasomal degradation (26).
However, these only represent a tip of the iceberg. Recent advances in systematic approaches have allowed us to unbiasedly uncover ISGs with previously uncharacterized antiviral activity. Compiled ISG libraries have facilitated focused loss-of-function or gain-of-function screens of hundreds of ISGs, illuminating the contribution of individual ISGs in varied viral contexts (27–30). Furthermore, as advances in omics approaches allow examination of cellular changes on a systemic level, attention is shifting to how ISGs interact and even synergize with one another (27, 31, 32).
Moreover, detailed mechanistic studies are still needed in order to unravel their mode of action. As ISGs may employ different antiviral mechanisms against different viruses, studies in varied viral systems will illuminate how ISGs might recruit different cellular pathways or factors.
Rather than provide a comprehensive, surface-level view of myriad ISGs with their arrayed antiviral functions, we have chosen to focus on a subset of ISGs that interfere with viral RNA processes. Recent studies have provided an emerging view on the diversity and complexity of RNA-based mechanisms by which different ISGs inhibit viruses with an RNA genome.
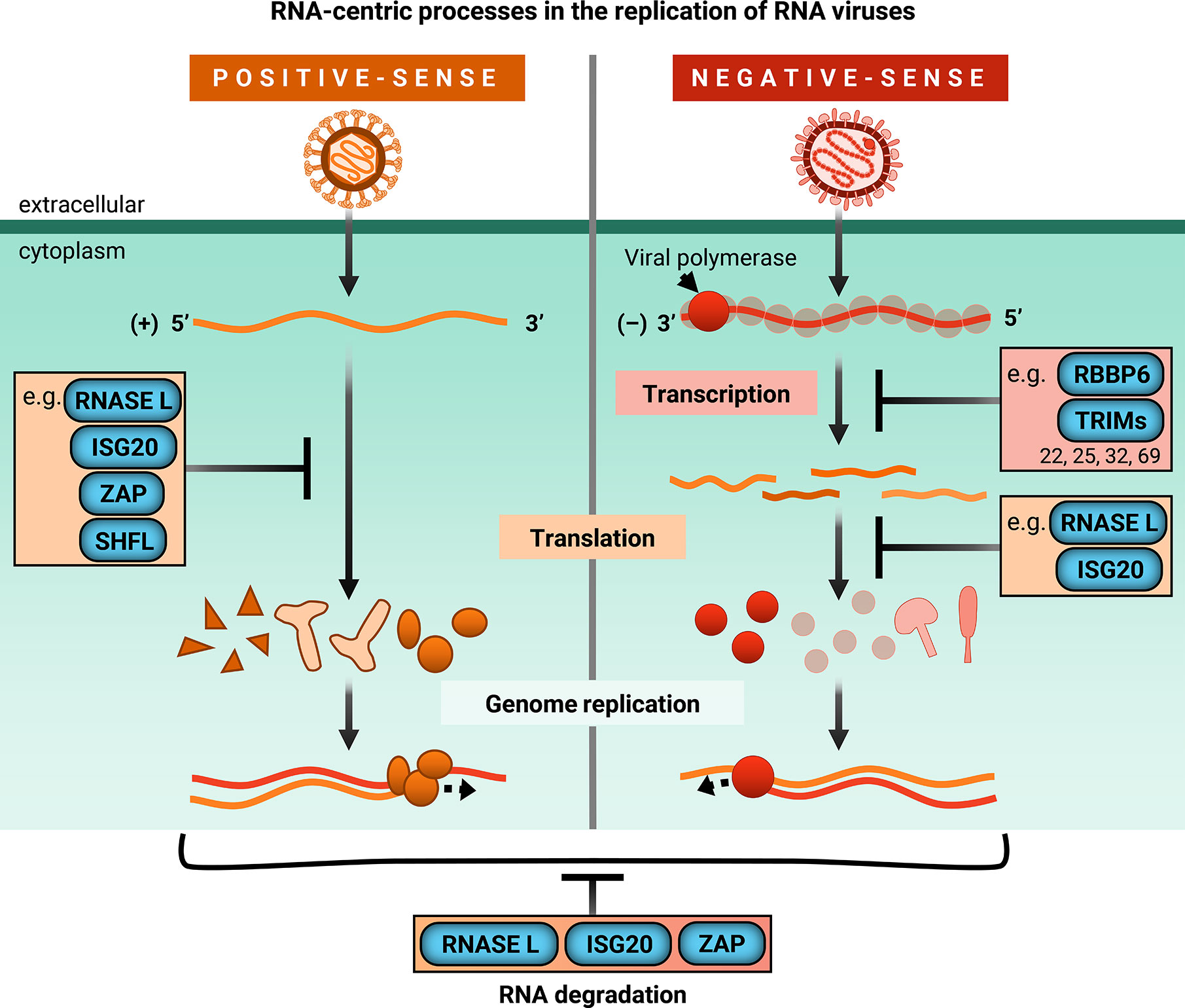
With the exception of retroviruses which replicate through a DNA intermediate, single-stranded RNA (ssRNA) viruses can generally be categorized as positive-sense and negative-sense. Positive-sense (+) ssRNA viruses possess genomes that generally resemble mRNA, in that it can be translated directly by host translation machinery. However, negative-sense (-) ssRNA viruses code their proteins in the reverse orientation.
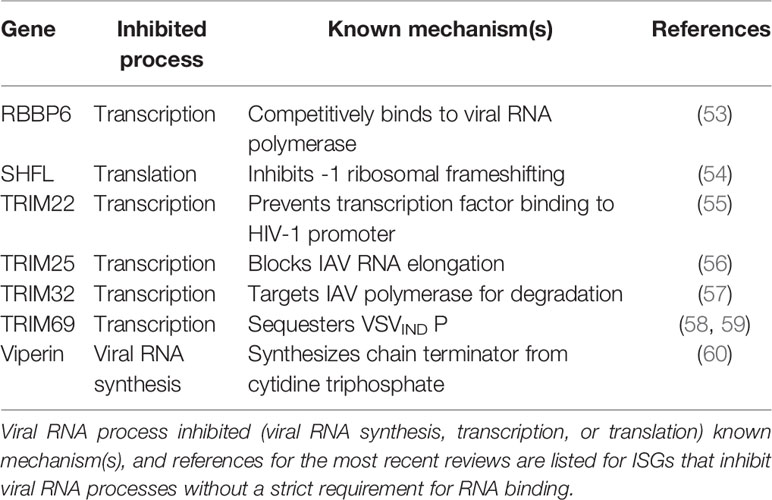
Therefore, they must package their own RNA-dependent RNA polymerases and transcribe their genomes before viral protein synthesis can occur. While there are many more ISGs that inhibit viral RNA processes (Tables 1 and 2), this review will highlight recent exciting work on a subset of ISGs that act in an RNA-centric manner to sense, degrade, or inhibit transcription or translation of both (+) and (-) ssRNA viral genomes (Figure 2).
We have chosen the ISGs here because at the time of writing of this review, they have not been comprehensively reviewed in the antiviral innate immunity field, and exciting developments have either illuminated nuances of well-characterized mechanisms or uncovered entirely new mechanisms by which these ISGs inhibit viral replication. We synthesized diverse antiviral mechanisms of individual ISGs and provided hypotheses for how cellular co-factors mediate the distinct antiviral activities of these ISGs, which not many previously published reviews have done. We will begin our discussion with the 2’-5’ oligoadenylate synthetase (OAS)/RNase L pathway, which both senses and degrades RNA, making it a potent early inhibitor of viral replication.
We will then segue into ISGs that both degrade RNA and inhibit translation, such as ISG20 and zinc finger antiviral protein (ZAP), before focusing on a novel means of translation inhibition by Shiftless (SHFL). We will end with a discussion of E3 ligases that inhibit viral transcription. Though there are many additional ISGs that block these RNA-centric steps of viral replication, this review focuses on ISGs that possess multiple or seemingly contradictory antiviral mechanisms. Protein-protein interactions or cellular co-factors could explain diverse antiviral mechanisms of individual ISGs.

reference link : https://www.frontiersin.org/articles/10.3389/fimmu.2020.605024/full



{kind=link}
[…] Two consecutive mutations (R203K/G204R) in the nucleocapsid (N) protein are associated… […]
[…] Two consecutive mutations (R203K/G204R) in the nucleocapsid (N) protein are associated… […]