










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


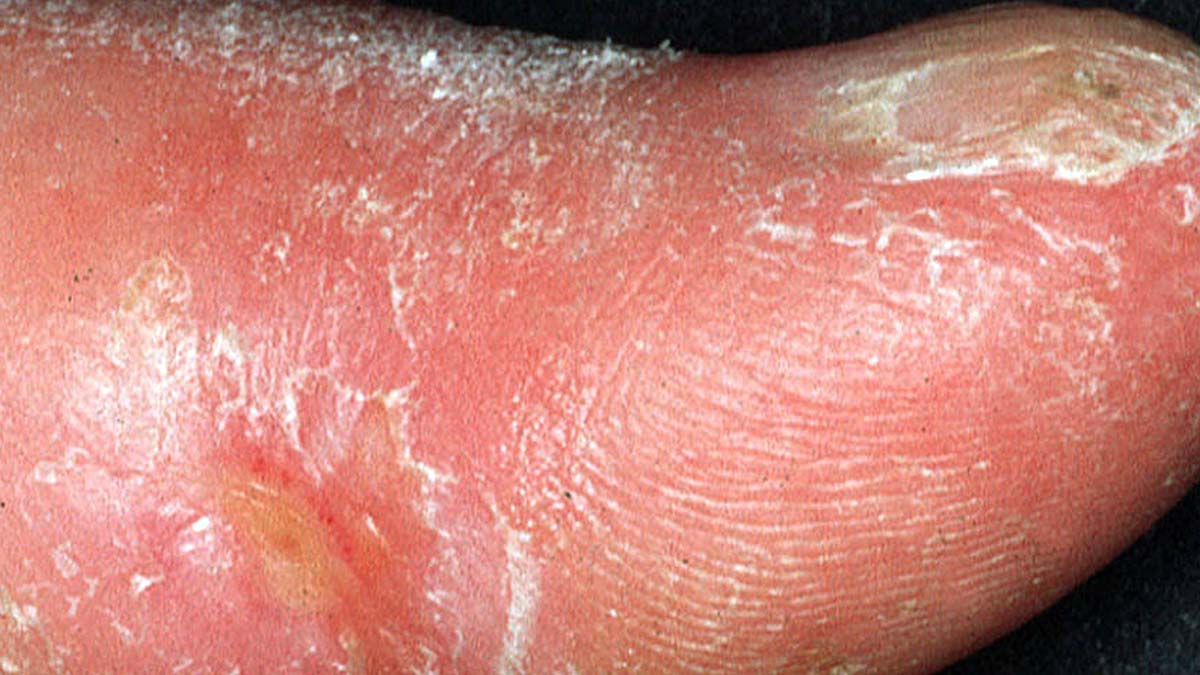
A subset of these cells, according to the study, may lie at the origins of scleroderma – a rare autoimmune disease. The findings open a new direction for developing a future therapy against this devastating, incurable disorder.
Scleroderma (from the Greek skleros, meaning “hard,” and derma, meaning “skin”) is characterized by the formation of an abnormally hard, inflexible layer of skin on the arms, legs and face. Its manifestations vary greatly among patients. In about a third of the cases, the disease, which mainly strikes women aged 30 to 50, advances rapidly and spreads beyond the extremities, causing life-threatening damage to internal organs.
Immune-regulating drugs that normally bring relief to people with autoimmune diseases are less effective in scleroderma, which has a higher mortality rate than other rheumatic disorders.
“Scleroderma is one of the most frustrating disorders to treat – we can alleviate some of the patient’s symptoms, but usually we cannot significantly affect the cause of the disease, block its progression or reverse its course,” says Prof. Chamutal Gur, a senior physician in the Rheumatology Department at Hadassah University Medical Center, who led the new study in Amit’s lab in Weizmann’s Immunology Department.
Her interest in this disease is not only professional: Two of her cousins have been diagnosed with scleroderma. When she joined Amit’s lab as a postdoctoral fellow some three years ago, her goal was to get to the root of this puzzling disease.
The study was conducted in collaboration with Dr. Hagit Peleg, Dr. Suhail Aamar, Dr. Fadi Kharouf, Dr. Anat Elazary and other rheumatologists at Hadassah University Medical Center, and with Prof. Alexandra Balbir-Gurman, who supervised the clinical aspect of the study, and Dr. Yolanda Braun-Moscovici, both of the Rambam Health Care Campus.
The researchers collected skin samples from nearly 100 scleroderma patients and from more than 50 healthy volunteers who served as a control group, in the largest-ever study of this kind to explore the disease. While perfecting the sample collection technique, Gur performed nearly 20 skin biopsies on herself.
As scleroderma is considered an autoimmune disorder—that is, one in which the immune system attacks the person’s own body—the researchers looked for immune cell differences between the control and patient groups. But contrary to what would be expected of an autoimmune disease, the analysis failed to reveal a characteristic, global pattern of immune abnormalities in most of the patients. Instead, to their surprise, the researchers found it were the patients’ fibroblasts that differed significantly from those of the controls.
Aside from roles in growth and wound healing, fibroblasts were thought to be mere “scaffolding” holding cells in place. The new study challenges this humdrum picture: The researchers found that fibroblasts can be divided into about ten major groups, each performing different and often vital functions, from conveying immune system signals to affecting metabolism, blood clotting and blood vessel formation. These groups can be further broken into some 200 subtypes.
Most importantly, the researchers managed to identify a subset of fibroblast whose concentration drops sharply in the early stages of scleroderma. They named these cells Scleroderma-Associated Fibroblasts, abbreviated as ScAFs (which is also short for “scaffold”). Whereas in healthy controls ScAFs accounted for nearly 30 percent of all fibroblasts, this percentage decreased dramatically in scleroderma patients and continued to plummet as the disease progressed.
They also identified biological markers correlated with specific kinds of organ damage; these markers can help physicians administer a personalized treatment, in order to prevent life-threatening complications. The research also revealed ScAF-related signaling pathways that can be targeted in future scleroderma therapies.
“The reduction in the size of a critical subset of fibroblasts appears to be an early event in the course of scleroderma,” Amit says. “It might be possible to design a therapy that will make up for this loss, slowing the progression of the disease.”
“Our approach is relevant to other diseases,” adds Dr. Shuang-Yin Wang of Amit’s lab, who led the study’s data analysis using artificial intelligence tools. “It reveals the enormous potential of meticulous tissue analysis involving advanced single-cell technologies for uncovering disease dynamics.”
“Integrating the latest single-cell genomic research techniques with clinical data can shed new light on diseases whose origins are currently obscure,” Amit concludes.
Tissue fibrosis is a pathological condition in which the diseased or damaged tissue is replaced by excessive extracellular matrix (ECM), leading to tissue hardening and loss of organ function. Secretion of ECM by activated fibroblasts (myofibroblasts) is a physiological response of the wound healing program.
However, unlike canonical dermal wound healing, in the context of fibrosis, fibroblasts remain persistently activated, and is likely sustained through positive feedback loops between fibroblast activation, inflammation (1), tissue stiffness (2), and vascular damage (3). When critical organs such as lungs, heart, kidney or liver become affected, the consequences of such over-scarring become fatal. For this reason, fibrosis is a major contributor to many diseases and ∼ 40% of mortalities worldwide are correlated with some form of underlying fibrosis (4).
Even though it poses a significant public health and socioeconomic burden, fibrosis still remains an irreversible condition with very limited treatment options, and this problem is compounded by the scarcity of biomarkers for disease susceptibility (5). Despite strong evidence for the pro-fibrotic activities of pathways such as TGF-β and endothelin (among others), clinical trials utilizing inhibitors of these signaling cascades have not lived up to the potential established in preclinical studies (4), (6).
One explanation for this discrepancy is the presence of redundant pathways that can eventually bypass the inhibition of a single node, given the multifactorial nature of fibrosis. Thus, there remains important gaps in the understanding of the molecular and cellular features that underlie this pathology. In this regard, preclinical animal models that recapitulate both the temporal and multifactorial nature of fibrosis is important to unravel the molecular underpinnings of this complex disease and illuminate potential new routes of therapeutic intervention.
An example of a fibrotic disorder with poor prognosis is scleroderma, a group of skin fibrotic disorders with different subtypes classified by the extent of tissue involvement. The subcategories of scleroderma are – Localised scleroderma (LoS) and Systemic Scleroderma or systemic sclerosis (SSc). LoS is limited to affecting only certain areas of the skin (7). On the other hand SSc is a progressive fibrotic disease with inflammatory, autoimmune and vascular defects (8).
Fibrosis characteristically initiates in the skin of distal extremities. The skin manifestations are limited to these areas in the case of limited SSc, however large areas of skin are involved in case of diffused SSc. Unlike, LoS, which negatively impacts the quality of life, SSc can progress to involve critical organs such as the lungs, heart and kidney often with fatal consequences. Many animal models have been developed to study SSc, and they have led to useful insights about the disease and represent different pathological and molecular subsets found in SSc (7) (9).
However, none of these models can be considered holistic in the sense that SSc encompasses fibrotic, inflammatory, autoimmune and vasculopathic components in the skin followed by progression of the disease to internal organs. Currently, most mouse models such as bleomycin treatment, TGF-β1, TSK2 and TSK1, uPAR mouse models lack at least one of these components (10).
The only mouse model that recapitulates all of these four aspects of SSc is the Fli1+/- Klf5+/- double heterozygote mouse model (11)(10). The Fli1+/- Klf5+/- model provides an excellent example of how creating a double haploinsufficient mice, which mimics the epigenetic suppression of Fli1 and Klf5 observed in SSc derived fibroblasts, can spontaneously lead to development of all cardinal defects of SSc followed by progression of fibrosis to the lungs. However, one drawback of such a global haploinsufficiency model would be the difficulty in distinguishing systemic effects from local ones.
Nevertheless, it is also important to characterize if these molecular and histological cross-species parallels can lead to a similar symptomatic course of disease development in the mouse. There is lacuna for mouse models which can faithfully reproduce the symptomatic pathogenesis along with the histological and molecular similarities to the human disease.
A mouse model which can manifest the earliest symptoms of the disease such as Raynaud’s like phenomenon and puffy fingers (both indicative of vascular defects of SSc), followed by chronic inflammation, systemic autoimmunity, progressive fibrosis in skin and involvement of other internal organs would be a valuable tool to increase our understanding of the etiology of SSc and illuminate new ways to treat this disease.
reference link : https://www.biorxiv.org/content/10.1101/2022.01.26.477822v1.full
More information: Chamutal Gur et al, LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma, Cell (2022). DOI: 10.1016/j.cell.2022.03.011



{kind=link}