








")


 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")


The study findings were published in the peer reviewed journal: Nature.
https://www.nature.com/articles/s41586-022-04802-1
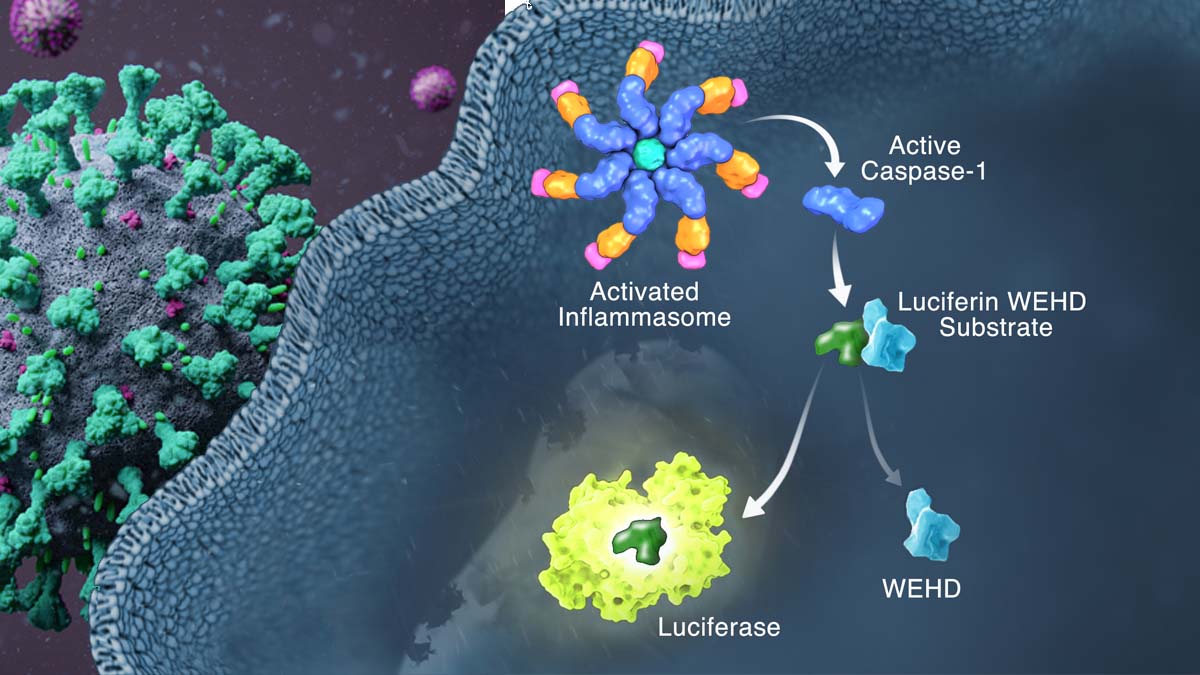
Severe COVID-19 is characterized by persistent lung inflammation, inflammatory cytokine production, viral RNA, and sustained interferon (IFN) response all of which are recapitulated and required for pathology in the SARS-CoV-2 infected MISTRG6-hACE2 humanized mouse model of COVID-19 with a human immune system 1–20.
Blocking either viral replication with Remdesivir 21–23 or the downstream IFN stimulated cascade with anti-IFNAR2 in vivo in the chronic stages of disease attenuated the overactive immune-inflammatory response, especially inflammatory macrophages.
Inflammasome activation and its accompanying inflammatory response is necessary for lung inflammation, as inhibition of the NLRP3 inflammasome pathway reverses chronic lung pathology.



{kind=link}