









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



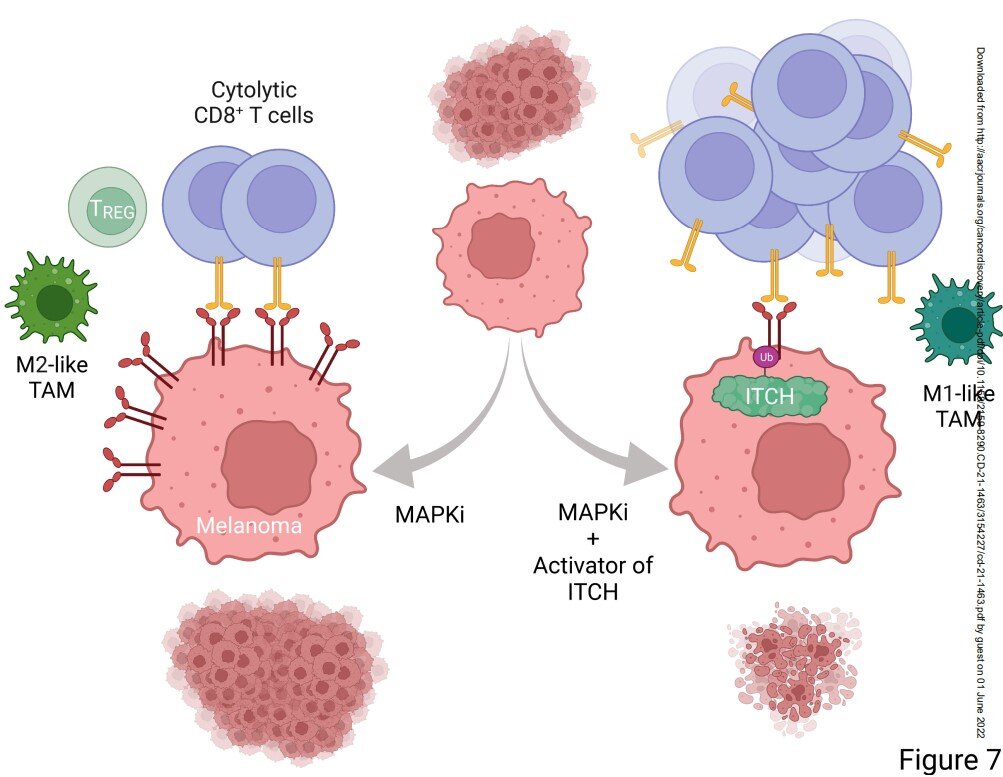
This approach, in combination with existing therapies, could improve treatment responses of metastatic melanoma and other cancers by suppressing resistance to current therapies.
Lo and his co-authors published their findings May 31 in the journal Cancer Discovery.
By searching a trove of small molecules at a National Institutes of Health library, they found and deployed a small molecule, which they characterized to be an ITCH activator. By activating ITCH, the small molecule degrades tumor cell-surface PD-L1. This small molecule, when used together with an existing therapy, suppresses relapses of melanoma in animal models.
Reducing the accumulation of PD-L1 clears the path for tumor-killing T-cells to do their work. “Once ITCH is activated, it’s now able to degrade or destabilize tumor surface PD-L1,” Lo said. “And once PD-L1 is degraded, then there are more T cells active to help therapies work better.”
Lo and his lab have been focusing on developing mutation-targeted therapy, as common cancer mutations drive disease progression by hyper-activating the so-called MAPK pathway. Therapy targeting the MAPK pathway for patients with metastatic cutaneous melanoma is associated with a high rate of response. However, the disease often comes back in a process called acquired resistance, causing clinical relapses.
In collaboration with another UCLA team led by James Wohlschlegel, Ph.D., professor of biological chemistry at the David Geffen School of Medicine, Yang identified ITCH as the protein that binds to surface PD-L1 and tags it biochemically for degradation by the tumor cell. In follow-up work, “We were excited to go further to find a potential path for this knowledge to help patients with cancers. Identification of a small molecule that can activate ITCH became a priority,” said Yan Wang, a first-year Ph.D. student who joined the Lo Lab from the department of molecular and medical pharmacology.
Programmed death-ligand 1 (PD-L1), with the gene name CD274, was first discovered in interleukin (IL)-3-deprived LyD9 (murine hematopoietic progenitor) and 2B4-11 (murine T cell hybridoma) cell lines in 1992(1) and was described as B7-H1 by Dong et al (2) in 1999. PD-L1 is the third member of the B7 family that does not bind CD28, cytotoxic T-lymphocyte A4 or inducible co-stimulator, and has 10-25% homology with B7.1 and B7.2 proteins (2). PD-L1 is encoded by the PDCDL1 gene, which was discovered at p24.1 on human chromosome 9.
The amino acid sequence of PD-L1 is encoded by 7 exons, which form a protein of ~40 kDa. PD-L1 is a type I transmembrane protein, is part of the immunoglobulin (Ig) superfamily and is composed of IgV-like and IgC-like extracellular domains, a hydrophobic transmembrane domain and a short cytoplasmic tail composed of 30 amino acids.
The engagement of PD-L1 and PD-1 on cancer cells activates Src homology region 2 domain-containing phosphatases, which inhibit the T cell receptor (TCR) pathway. Inhibition of the TCR pathway leads to inhibition of T cell activities, including proliferation, survival and cytokine production, such as that of IL-2, tumour necrosis factor α (TNF-α) and interferon γ (IFN-γ) (5), as well as the inhibition of B7-1 and T cell tolerance (6,7).
To the best of our knowledge, the present review will discuss for the first time how autophagy, a protein degradation pathway that regulates homeostasis of cells, also regulates PD-L1 expression on cancer cells.
Discussion
T cell immunity is essential for homeostasis of the body, as it identifies antigens and kills cells with gene mutations or with aberrant pathology, which includes tumour cells. Unfortunately, excessive activation of uncontrolled T cells can also kill normal tissue cells, contributing to autoimmune diseases such as rheumatoid arthritis (51).
Therefore, preventing autoimmunity by regulating activated T cells is an important feature of immune homeostasis. The co-inhibitory immune checkpoints, which include CTLA4-CD80, PD-1-PD-L1, galectin-9-T cell immunoglobulin mucin-3 and TCR-lymphocyte activation gene 3, can regulate the activity of T cells under normal physiological conditions (52).
However, upregulation of these inhibitory checkpoints leads to the immune microenvironment becoming immunosuppressed (53), which can cause immune tolerance and immune escape. The PD-1-PDL1 axis has been particularly identified as the most clinically significant, as antibodies against it have led to benefits in a variety of cancer types such as NSCLC, melanoma and gastric cancer (9,11,17). Inhibition of PD-L1 expression on tumour cells heightens immunosurveillance and decreases the immune checkpoint function derived from PD-L1(5). Hence, it is critical to explore the mechanisms that regulate the expression of PD-L1.
In the past ten years, the mechanisms that regulate PD-L1 expression via different pathways have been explored. First, the genomic alternation/rearrangements in chromosome 9p24.1, on which CD274 is located, have been identified to upregulate PD-L1 expression (54-58). It has been reported in the literature that amplification and mutations in the Janus kinase (JAK) family promote the upregulation of PD-L1 expression by inducing its mRNA expression (55,59).
The increase in activity of the JAK2/STAT signalling pathway resulting from gene mutations also increases PD-L1 expression (55,59). DNA double-strand breaks also upregulate PD-L1 expression by activating the STAT signalling pathway via the kinases ATM/ATM and Rad3-related/checkpoint kinase 1 (60,61). The expression of PD-L1 was induced by disrupting the CD274 3’UTR as well, by using CRISPR technology/Cas9 or miRNAs, such as miR-200, miR-34a, miR-152 and miR-424 (62-65).
Epigenetic regulation, histone acetylation and methylation boost PD-L1 expression on melanoma and pancreatic cancer cells (66-68). In addition, oncogenic transcription factors, for example MYC, can combine with the PD-L1 promoter to enhance PD-L1 expression in hepatocellular carcinoma, human melanoma and NSCLC cell lines (69). Anaplastic lymphoma kinase can promote PD-L1 expression via STAT3(70).
Besides MYC and ALK, the mutated and amplified HIF1/2α (hypoxia-inducible factor 1/2-α), NF-κB, phosphatase and tensin homologue/PI3K, mitogen-activated protein kinase and epidermal growth factor receptor oncogenic pathways can upregulate PD-L1 mRNA expression on melanoma, ovarian cancers and lung squamous carcinoma cells (71-76). In addition, the IFN-γ/JAK/STAT1 inflammatory pathway is used by cancer cells to enhance PD-L1 mRNA expression (77,78).
Several inflammatory cytokines, such as Toll-like receptor 3, TNF-α, transforming growth factor β, IFN-α/β and IL-4/6/17/27 have been demonstrated to upregulate PD-L1 mRNA expression on tumour cells or tumour-associated stromal cells (78-85). Post-transcriptional modifications, such as N-linked glycosylation (86), serine/threonine and tyrosine phosphorylation (87), polyubiquitination (88) and palmitoylation (89) have been found to serve significant roles in protein stability, as well as PD-L1 translocation regulation.
The present study demonstrated that autophagy can be induced by various molecules to downregulate PD-L1 through NF-kB, STAT3, HDAC6 or the degradation of autophagic flux. Whether autophagy can regulate PD-L1 expression through MYC, ALK, HIF1/2, several inflammatory cytokines, or other mechanisms remains to be investigated. Oncogenic pathways, such as PI3K/AKT and Ras/Raf/MEK/ERK, can induce autophagy and also regulate PD-L1 expression, which suggests that these pathways may regulate PD-L1 expression via autophagy.
mTOR is a major negative regulator of autophagy (90), which can be inhibited by RAP to reduce the expression of PD-L1(91). When the PI3K/AKT signalling pathway, as the main modulator upstream of mTORC1(92) is activated, this can phosphorylate tuberous sclerosis complex 2 to abolish the formation of the TSC1/2 complex, hence activating mTOR to inhibit autophagy (93). When the PI3K-AKT pathway is activated by epidermal growth factor and IFN-γ, it induces PD-L1 expression (91,94,95). Furthermore, the PI3K-AKT pathway also can regulate PD-L1 expression in the absence of IFN-γ in various cancer types, such as NSCLC, CRC, glioma, breast cancer and melanoma cells (72,76,91,94,96), which suggests that this regulation occurs, at least in part, by changing the mRNA expression of PD-L1 (74,97).
In addition, the MEK-ERK signalling pathway has been identified frequently to be activated in a variety of cancer types such as hepatocellular carcinoma and colon cancer (98,99). MEK-ERK is located on the outer surface of autophagosomes and promotes the production of Beclin1 protein by inducing the lipidation of LC3-I to LC3-II, hence enhancing autophagy (100). In addition, the MEK/ERK module promotes autophagy via the AMPK-MEK/ERK-TSC-mTOR signalling pathway (101).
Continuous activation of the Ras/Raf/MEK/ERK pathway may enhance the autophagy flux in cells, and mRNA levels of LC3B and SQSTM1 are also increased (102). When the MEK-ERK pathway was inhibited by chemical or genetic inhibitors, PD-L1 transcription induced by IFN-γ was inhibited in multiple myeloma cells (103).
In concert with this, when the MEK-ERK signalling pathway was activated by phorbol myristate acetate, PD-L1 expression was enhanced. By contrast, when MEK was inhibited in tumours, PD-L1 expression was decreased in mouse-derived breast cancer cell lines (103,104). Inhibition of the MEK-ERK signalling pathway abrogates increased PD-L1 expression stimulated by TLR ligands, which has been observed in various cancer cells and antigen presenting cells, such as myeloma, bladder cancer, lymphoma and dendritic cells (94,97,98,103,105-107).
Higher mutation rates of RAS and excessive activation of the RAS pathway have been demonstrated to increase PD-L1 expression among human lung and colorectal tumours (108). Besides, autophagy, as a pro-survival tumour mechanism, can be mediated by PD-L1 to escape the immune system. It has been reported that PD-L1 induces autophagy via mTORC signalling to promote the proliferation of ovarian cancer cells (109).
As aforementioned, activated autophagy may reversely upregulate the PD-L1 expression through the ATG/autophagy/FOXO3A/miR-145 axis, forming a positive feedback loop to create a favourable environment for tumour progression.
As discussed above, the mechanisms that autophagy uses to regulate PD-L1 expression were reviewed. Autophagy can both positively and negatively regulate the PD-L1 expression on cancer cells. However, whether oncogenic pathways, such as PI3K/AKT and Ras/Raf/MEK/ERK, regulate PD-L1 expression via autophagy needs to be further elucidated. In addition, the mechanism by which autophagy effects PD-L1 expression remains to be further clarified in future studies.
In conclusion, autophagy can regulate PD-L1 expression in a number of cancer types via various mechanisms. Joint use of autophagy regulators and drugs targeting the PD-1/PD-L1 axis may enhance the therapeutic effect, hence improving the prognosis of patients with cancer.
REFERENCE LINK : https://www.spandidos-publications.com/10.3892/br.2021.1460
More information: Zhentao Yang et al, Enhancing PD-L1 Degradation by ITCH during MAPK Inhibitor Therapy Suppresses Acquired Resistance, Cancer Discovery (2022). DOI: 10.1158/2159-8290.CD-21-1463. aacrjournals.org/cancerdiscove … -by-ITCH-during-MAPK



{kind=link}