








")


 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



La vitamina K è un gruppo di vitamine liposolubili essenziali per la coagulazione del sangue e la salute delle ossa. È stato suggerito che la vitamina K possa anche avere proprietà antiossidanti e antinfiammatorie, che potrebbero potenzialmente aiutare a prevenire la ferroptosi e mitigare i danni causati dall’infezione da SARS-CoV-2.
Alcuni studi hanno studiato la relazione tra lo stato della vitamina K e gli esiti di COVID-19. Ad esempio, uno studio pubblicato sull’American Journal of Clinical Nutrition nel 2021 ha rilevato che bassi livelli di vitamina K erano associati a un aumento del rischio di COVID-19 grave e mortalità.
I ricercatori del Dipartimento di biologia medica e biochimica, Facoltà di medicina, Università Nicolaus Copernicus di Toruń-Polonia hanno condotto una revisione dello studio esaminando il potenziale della vitamina K nella soppressione della ferroptosi indotta da SARS-CoV-2.
collegamento di riferimento: https://www.mdpi.com/2076-3921/12/3/733

La malattia da coronavirus 2019 (COVID-19), causata dalla sindrome respiratoria acuta grave coronavirus 2 (SARS-CoV-2), continua a rappresentare una sfida significativa sia dal punto di vista medico che sociale [1]. I primi casi di infezione da SARS-CoV-2 sono stati osservati alla fine del 2019 a Wuhan, la capitale della provincia di Hubei, in Cina [2].
Tuttavia, molti altri sintomi, come perdita del gusto o dell’olfatto, mal di gola, congestione o naso che cola, mal di testa, dolori muscolari o muscolari, nausea o vomito e diarrea, sono stati riportati in individui con COVID-19 [1,2] . L’infezione causata da SARS-CoV-2 è stata associata ad un aumentato rischio di coagulopatie [5]. I disturbi dell’emostasi possono portare a coagulopatia intravascolare disseminata (DIC) e grave emorragia da ictus [5].
Uno dei fattori che regolano il funzionamento del sistema di coagulazione è la vitamina K [6]. Il risultato della carenza di vitamina K può essere facile formazione di lividi e sanguinamento spontaneo in caso di carenze significative [7]. Finora, la vitamina K è stata associata principalmente alla regolazione del processo di coagulazione e all’omeostasi del sistema scheletrico.
Gli ultimi rapporti scientifici indicano che la vitamina K ha proprietà antiossidanti [8]. Molte condizioni patologiche, comprese le malattie infettive, portano allo squilibrio redox. Durante il COVID-19 si osserva anche un aumento della generazione di specie reattive dell’ossigeno (ROS) [9].
Lo stress ossidativo può essere correlato sia alla gravità della malattia e ai suoi sintomi, sia alla prognosi e alla sindrome post-COVID-19 [10].
Una delle strategie terapeutiche per ridurre la ferroptosi nel corso di COVID-19 potrebbe essere l’integrazione con vitamina K grazie alle sue proprietà antiossidanti. La vitamina K, in quanto molecola lipofila, può ridurre la formazione di LOOH.
Questo articolo mira a indicare la relazione tra l’aumento dello stress ossidativo nel corso di COVID-19, la ferroptosi e il potenziale uso terapeutico della vitamina K per ridurre l’impatto negativo dei ROS sulle cellule. Questa pubblicazione è un’analisi della letteratura scientifica più recente dai database PubMed, Google Scholar e Web of Science.
Ossidante – Equilibrio antiossidante e perossidazione lipidica
La conseguenza del consumo di ossigeno per vivere è la generazione di ROS nelle cellule. Il termine ROS comprende sia i radicali liberi dell’ossigeno che le specie non radicaliche. I principali ROS includono, tra gli altri, il radicale anione superossido (O 2 •− ), il perossido di idrogeno (H 2 O 2 ), il radicale ossidrile (HO • ) e l’ossido nitrico ( • NO), che è anche una specie reattiva dell’azoto (RNS ) [13].
I ROS sono generati durante il normale funzionamento delle cellule [14]. Le loro principali fonti cellulari sono i mitocondri e le NADPH ossidasi (NOX) [15,16]. Se l’equilibrio tra generazione di ROS e meccanismi di difesa antiossidante è distorto a favore degli ossidanti, questa condizione porta a quello che è noto come stress ossidativo [17,18], la cui potenziale conseguenza può essere il danno cellulare a livello molecolare [19], compresa l’ossidazione di proteine, DNA e lipidi [20].
È stato dimostrato che lo stress ossidativo svolge un ruolo essenziale nella patogenesi di numerose malattie [21,22]. Tuttavia, i ROS hanno anche un ruolo nella regolazione delle vie di segnalazione, influenzando il corso dei processi biologici [20,23]. Secondo l’ultimo aggiornamento del concetto di stress ossidativo, viene fatta una distinzione tra stress ossidativo quando un’elevata generazione di ROS porta a danno molecolare e stress ossidativo quando i ROS si verificano a livelli fisiologici, svolgendo un ruolo importante nella segnalazione redox [24]. I fattori chiave nella segnalazione redox sono considerati O 2 •− e H 2 O 2 . La loro generazione avviene sotto il controllo di fattori di crescita e citochine da parte di più di 40 enzimi [24].
Uno dei processi responsabili dello stress ossidativo è la perossidazione lipidica (LPO) [25]. Nel corso di questo processo distinguiamo tre fasi: inizio, propagazione e conclusione [26,27]. Il ciclo inizia con il distacco di un atomo di idrogeno da una molecola del gruppo metilenico degli acidi grassi polinsaturi (PUFA) e la formazione di un radicale libero lipidico (L • ) . Quindi, si verifica il riarrangiamento dei doppi legami e la formazione di un radicale più stabile contenente dieni coniugati (CD) [28,29,30].
Il fattore scatenante della LPO può essere, tra gli altri, HO • e un radicale idroperossilico (HO 2 • ) [26,31]. Durante la fase di propagazione, il radicale lipidico reagisce con l’ossigeno, formando un radicale perossilico (LOO • ). È un radicale ad alta energia che può staccare un atomo di idrogeno da una molecola lipidica e trasformarsi in una molecola LOOH.
Questa reazione è accompagnata dalla formazione di un altro L • . I LOOH, formati durante la fase di propagazione, sono i principali prodotti primari del processo LPO [26]. Possono quindi subire una riduzione di uno o due elettroni. Durante la riduzione e l’ossigenazione di un elettrone mediate dal ferro (secondo l’equazione: LOOH + Fe 2+ → O 2 → OLOO • + OH + Fe 3+ ), si formano radicali perossilici epossialilici (OLOO • ), che causano ulteriori radicali liberi perossidazione di catena mediata [32].
Nella fase di terminazione, i radicali liberi (L • , LOO • ) formati reagiscono tra loro, creando prodotti che non sono radicali liberi [28]. Le trasformazioni finali dei prodotti LPO, durante la β-eliminazione e la decomposizione dei derivati PUFA, portano alla formazione di prodotti secondari del processo di perossidazione, tra gli altri, malondialdeide (MDA), 4-idrossinonenale (4-HNE) e isoprostani [29,33 ]. I prodotti LPO, inclusi LOOH e aldeidi, sono altamente reattivi, possono causare danni alle proteine e al DNA e causare alterazioni selettive nella segnalazione cellulare [34].
L’ossidazione dei PUFA può anche avvenire con la partecipazione di enzimi, tra gli altri, lipossigenasi (LOX) e cicloossigenasi (COX) [35]. È stato dimostrato che la produzione di LOOH nelle membrane delle cellule neoplastiche, con la partecipazione di LOX, potenzia la ferroptosi indotta da erastina (un inibitore dell’antiportatore di cistina/glutammato) e RSL3 (un inibitore della glutatione perossidasi 4 (GPx4)) [36].
Il sistema di difesa antiossidante presente nelle cellule, responsabile della rimozione dei ROS, è formato da enzimi e antiossidanti non enzimatici. La barriera enzimatica comprende, tra gli altri, superossido dismutasi (SOD), GPx, catalasi (CAT) e tioredossina (Trx).
Gli scavenger non enzimatici di ROS includono vitamine o loro analoghi (tra gli altri, vitamine C, A, E; flavonoidi, coenzima Q10 (CoQ10)), metaboliti (p. es., melatonina, bilirubina) e minerali (p. es., zinco, selenio) [37 ]. Una funzione antiossidante nelle membrane è svolta anche dalla vitamina K [38].
Il sistema Trx e il sistema antiossidante del glutatione sono i due principali sistemi antiossidanti tiolo-dipendenti nei corpi dei mammiferi [39]. Il sistema Trx, che include Trx, tioredossina reduttasi (TrxR), tioredossina perossidasi (TPx) e NADPH, partecipa alla regolazione dell’espressione genica e alla modulazione delle vie di segnalazione cellulare, ma anche alla disintossicazione dei LOOH [40]. La SOD neutralizza l’O 2 •− mediante la sua dismutazione in perossido di idrogeno e ossigeno.
Nei mammiferi sono state identificate tre isoforme SOD: isoforma CuZn (CuZn-SOD o SOD1) presente nel citosol e nel nucleo; SOD di manganese (Mn-SOD o SOD2) localizzata nei mitocondri; e CuZn-SOD extracellulare (EC-SOD o SOD3) [41,42]. CAT è presente principalmente nei perossisomi [14]. Contiene una molecola di ione ferrico nel suo sito attivo e conduce la dismutazione di H 2 O 2 in ossigeno e acqua. Oltre a questa funzione di base, CAT partecipa anche alla decomposizione, tra gli altri, di idroperossidi, metanolo, etanolo, azide e formiato [43,44]. Alcuni studi confermano che il CAT può decomporre il perossinitrito [45] e ossidare l’ossido nitrico in nitrito [43,44].
La GPx è anche coinvolta nella rimozione di H 2 O 2 e LOOH [46,47], secondo le seguenti reazioni [48]:
H 2 O 2 + 2GSH → 2H 2 O + GSSG (1) e LOOH + 2GSH → LOH + GSSG + H 2 O (2) (GSH—glutatione ridotto, GSSG—glutatione ossidato).
Ad oggi, negli esseri umani sono stati rilevati otto diversi GPx. In cinque di essi (GPx1-4 e GPx6), si verifica un residuo di selenocisteina nel sito attivo [49]. Gli enzimi della famiglia GPx, in particolare GPx4, sono importanti regolatori dei LOOH nelle cellule. Catalizzano la riduzione a due elettroni di LOOH portando alla formazione di alcol redox-inerte (2) [50]. Il co-substrato utilizzato durante la riduzione del LOOH ad alcol è il GSH [51]. L’attività GPx4 viene rilevata nel citosol, nel nucleo cellulare e nei mitocondri. Questo enzima è considerato un regolatore chiave della ferroptosi. GPx4 può ridurre i lipidi complessi perossidati e silenzia anche LOX [49]. Detossificando gli idroperossidi nei lipidi di membrana, GPx4 riduce il grado di danno alla funzione di membrana e previene la generazione di prodotti reattivi del processo di perossidazione [52].
Meccanismi della ferroptosi e implicazioni sulla salute
Nel 2012, Brent Stockwell con i suoi colleghi, tra cui Scott Dixon, ha proposto il concetto della forma dipendente dal ferro della morte cellulare regolata, che è correlata all’ossidazione, definita ferroptosi, che è distinta dall’apoptosi, dalla necrosi non regolata e dalla necroptosi (necrosi regolata ).
Le principali caratteristiche morfologiche della ferroptosi sono il restringimento mitocondriale accompagnato da una maggiore densità della membrana mitocondriale e una membrana mitocondriale interna degenerata (cioè catena di trasporto degli elettroni) senza cambiamenti nel nucleo [53]. La condizione per la ferroptosi è l’ossidazione dei lipidi di membrana (in particolare il reticolo endoplasmatico, ER) e l’interruzione dei meccanismi che impediscono l’accumulo di lipidi ossidati. Pertanto, la ferroptosi non dovrebbe essere considerata un tipo di stress ossidativo ma piuttosto un accumulo di LOOH letali localizzati nelle membrane cellulari [54] (vedi Figura 1).
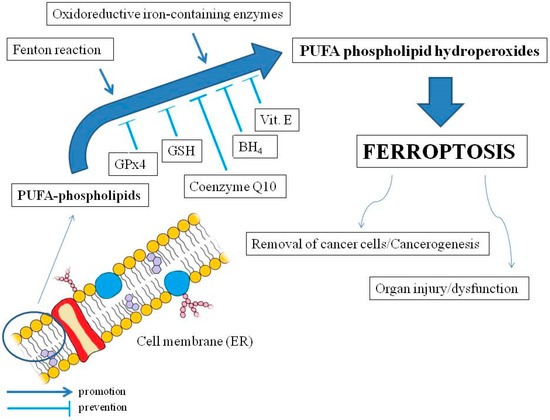
Figura 1. I principali meccanismi della ferroptosi e gli effetti sull’organismo umano. Gli acidi grassi polinsaturi (PUFA) nei fosfolipidi di membrana (principalmente reticolo endoplasmatico, ER) sono ossidati da specie reattive dell’ossigeno dalla reazione di Fenton o come risultato dell’attività di enzimi ossidoriduttivi contenenti ferro. L’ossidazione può portare alla ferroptosi per accumulo di idroperossidi fosfolipidici PUFA, che possono essere inibiti dalla glutatione perossidasi 4 (GPx4) e dal glutatione (GSH) solubili, nonché dai protettori della membrana lipofila come il coenzima Q10, la tetraidrobiopterina (BH4) e la vitamina E La ferroptosi può essere implicata nella morte delle cellule tumorali, ma può anche promuovere la cancerogenesi, nonché lesioni e disfunzioni d’organo (diagramma della membrana cellulare leggermente modificato, per gentile concessione di Holly Barker/The Science Hive).
Eccezionalmente implicati nella ferroptosi sono i fosfolipidi (PL) caratterizzati da un alto contenuto di PUFA. La formazione di idroperossidi PUFA in complessi con fosfolipidi (PLs-OOH) promuove la ferroptosi [54]. Tuttavia, non tutti i PL contribuiscono allo stesso modo alla ferroptosi.
Le fosfatidiletanolammine con una coda acilica grassa derivata dall’acido arachidonico o dall’acido adrenico (acido docosatetraenoico) sono più strettamente correlate alla ferroptosi rispetto ad altri PL [55]. Occasionalmente si osservano anche PL con due code PUFA. Sono più suscettibili alla ferroptosi [56]. È stato anche riscontrato che i PUFA liberi probabilmente non influenzano la ferroptosi [57], analogamente agli idroperossidi PUFA, ad esempio, dopo la loro scissione dai PL da parte della fosfolipasi A2β (iPLA2β) indipendente dal Ca 2+ [58]. A sua volta, l’incorporazione di acidi grassi monoinsaturi (MUFA) nei PL comporta un effetto anti-ferroptotico [59], e gli acidi grassi saturi in complesso con i PL non hanno alcun impatto su questo fenomeno [54].
L’ossidazione dei PL nella ferroptosi è associata alla transizione degli ioni ferro nello stato di ossidazione II (Fe 2+ ), che si traduce in una reazione di Fenton e nella produzione di ROS, nonché con enzimi contenenti ferro, come i LOX [60] ( generalmente richiedono Fe 3+ per l’attività [61]) o, ad esempio, ossidoreduttasi del citocromo P450 [62] (vedi Figura 1). Per quanto riguarda gli organelli cellulari, i mitocondri possono avviare o amplificare la ferroptosi indotta dalla carenza di cisteina associata alla deplezione di GSH.
Inoltre, questi organelli svolgono un ruolo centrale nel metabolismo ossidativo. La perdita di elettroni dalla catena di trasporto degli elettroni mitocondriale si manifesta con la formazione di O 2 •− e H 2 O 2 , che possono quindi reagire con Fe 2+ per guidare la chimica di Fenton e LPO [54]. Nel frattempo, come già accennato, la relazione tra ferroptosi e cambiamenti morfologici significativi della cresta mitocondriale è un dato di fatto [53].
Inoltre, l’inibizione della catena di trasporto degli elettroni mitocondriali attenua anche la ferroptosi indotta dalla fame di cisteina, così come l’esaurimento dei mitocondri [54]. LPO, oltre alle sue implicazioni nella ferroptosi, può essere un collegamento con altre forme di morte cellulare regolata, in particolare l’apoptosi e la morte dipendente dall’autofagia, ma anche: piroptosi, necroptosi, parthanatos e morte cellulare netotica [63].
L’abbondanza di ferro nella cellula dipende, a sua volta, dal suo trasporto con il sangue dall’intestino. Ciò coinvolge principalmente la transferrina (TF) e i suoi recettori cellulari [64]. La disponibilità cellulare di ferro, a sua volta, è regolata dalla ferritina (FER), che la immagazzina sotto forma di Fe 3+ e previene la ferroptosi [65].
Alcune proteine possono agire sulla ferroptosi influenzando la concentrazione di FER. Ad esempio, la chinasi atassia telangiectasia inibisce la sintesi di FER e quindi può promuovere la ferroptosi [66]. Altri meccanismi per la regolazione del pool di ferro cellulare e della ferroptosi sono correlati all’esportazione di ferro (ferroportina (FPN) ed esosomi) [67].
La disponibilità di Fe 2+ nella cellula è anche associata a composti a basso peso molecolare, incluso il GSH. Lo stoccaggio del ferro in FER richiede la formazione di un complesso GSH-ferro [68]. Pertanto, l’esaurimento del GSH può promuovere la ferroptosi non solo facilitando la produzione di ROS nella reazione di Fenton, ma anche inibendo la GPx4, che è particolarmente coinvolta nella prevenzione dell’LPO.
GSH e GPx4 sono considerati i principali inibitori della ferroptosi [69]. È interessante notare che la degradazione della GPx4, ad esempio attraverso la proteina ferroptosis-inducer-56 (FIN56) [70] o l’autofagia mediata da chaperone [71], è più favorevole alla ferroptosi rispetto all’inibizione dell’enzima. La carenza di GSH e quindi la promozione della ferroptosi possono anche derivare dall’inibizione della produzione di GSH, ad esempio, a causa dell’inibizione della glutammato-cisteina ligasi [72] o del blocco dell’assorbimento della cistina nel sistema di membrana Xc −, un sistema antiportatore di cistina/glutammato, ma anche come risultato dell’efflusso di GSH [69] o dell’aumento del catabolismo della cisteina (diossigenasi 1, CDO1) [73]. Inoltre, è stato riportato che la citata ligasi ha aumentato la resistenza cellulare alla ferroptosi non solo attraverso il contributo alla sintesi di GSH, ma anche come risultato della conversione del glutammato in peptidi γ-glutamil, il che suggerisce che il glutammato può promuovere la ferroptosi [72].
Importanti soppressori endogeni della ferroptosi sono anche antiossidanti lipofili, in particolare il CoQ10. È presente in diverse membrane cellulari, compresi i mitocondri, e protegge fortemente contro LPO. Dopo la lipoperossidazione, può essere recuperato con NADPH [74]. La soppressione della LPO e, quindi, della ferroptosi è anche associata a un altro antiossidante lipofilo endogeno, la tetraidrobiopterina (BH 4 ) [56]. Anche gli antiossidanti esogeni, ad esempio ingredienti dietetici come la vitamina E, possono proteggere dalla ferroptosi [75]. Inoltre, la ferroptosi può essere inibita dalla diidroorotato deidrogenasi (DHODH), riduttore del CoQ10 nei mitocondri [76], e dall’interleuchina-4-indotta-1 (IL4I1) attraverso la sintesi dell’indolo-3-piruvato (In3Py) [77].
La ferroptosi è implicata in molti fenomeni nell’organismo umano (vedi Figura 1). Ad esempio, l’attenuazione della ferroptosi nelle cellule T CD4 + con una dieta ricca di selenio (la selenocisteina si verifica nel sito attivo di GPx4) ha innescato un aumento del numero di cellule B di memoria e un’immunità virale di lunga durata [78].
Uno studio in vitro ha dimostrato che l’eccessivo assorbimento di PUFA da parte delle cellule tumorali in un ambiente acido può portare alla loro ferroptosi, mentre una dieta ricca di PUFA promuove la ferroptosi delle cellule tumorali nei topi [79]. A loro volta, le cellule T CD8 + uccidono le cellule tumorali attraverso la ferroptosi rilasciando interferone gamma (IFN-γ) e acido arachidonico, che si traduce in un aumento dell’incorporazione di PUFA nei PL [80], mentre una dieta ricca di colesterolo ha favorito un aumento L’assorbimento dei PUFA da parte delle cellule T CD8 + e la loro morte per ferroptosi, che possono promuovere la progressione del cancro [81].
Inoltre, la ferroptosi può essere implicata nella sindrome da disfunzione multiorgano o nel danno d’organo come conseguenza del sovraccarico di ferro o anche per altri motivi (segnati tra parentesi), ad es. cervello (malattie neurodegenerative), cuore, fegato (infezione da epatite C), reni e polmoni ( infezioni da Pseudomonas aeruginosa , SARS-CoV-2). La relazione tra ferroptosi e infiammazione non è altrimenti chiara [54].
I meccanismi regolatori descritti della ferroptosi (LPO causata da stress ossidativo mediato dal ferro e via di inibizione GPx4-GSH) possono essere cruciali per il trattamento di COVID-19 [11,12], compresa la sindrome post COVID-19 [10], poiché la condizione patologica nelle infezioni da SARS-CoV-2 è strettamente associata a disfunzione mitocondriale, sovraccarico di ferro, stress ossidativo e infiammazione [9,10,11,12].
Ferroptosi e COVID-19
COVID-19 si manifesta in molte complicanze così come alterazioni fisiologiche e biochimiche. Questi includono, tra gli altri, un aumento delle concentrazioni di cellule proinfiammatorie CD4 + T e CD8 + T, un massiccio rilascio di citochine (la cosiddetta tempesta di citochine), un aumento dello stato di coagulazione, danni all’emoglobina e disregolazione dell’omeostasi del ferro, compreso il sovraccarico di ferro, che è probabilmente un fattore significativo nella patogenesi del COVID-19 [82].
La ferroptosi non regolata può in parte spiegare la degenerazione cellulare e il danno tissutale della patogenesi della malattia polmonare [83]. Pertanto, le interazioni molecolari tra la regolazione sistemica e cellulare del ferro e il processo infiammatorio rappresentano una nuova area di ricerca cruciale per comprendere la patogenesi di COVID-19.
Il mantenimento dell’omeostasi del ferro è fondamentale per l’organismo. È interessante notare che sia i livelli di ferro bassi che quelli alti aumentano il rischio di infezione. In oltre il 75% dei pazienti critici sono stati osservati valori elevati di FER sierico, bassi livelli di ferro sierico e bassi livelli di transferrina entro tre giorni dal ricovero in unità di terapia intensiva, indicando l’importanza del ferro e delle proteine correlate [84].
In risposta all’infezione da SARS-CoV-2, in molti pazienti è stata osservata una disfunzione del metabolismo del ferro. L’aumento del metabolismo cellulare delle cellule ospiti e livelli di ferro ottimali sono necessari per la replicazione virale [85]. L’asse epcidina/ferroportina regola significativamente il ferro sistematico [86,87].
L’epcidina, in un’espressione regolare, ha una funzione protettiva nei confronti delle infezioni. Tuttavia, la sua sovraespressione in COVID-19 può essere dannosa e associata a vari sintomi della malattia e una prognosi peggiore [88,89]. L’epcidina essenzialmente sottoregola la FPN e quindi determina ipoferremia e sequestro di ferro a livello cellulare [86]. A causa dell’elevata concentrazione di ferro all’interno della cellula, il ferro cellulare libero (Fe 3+ ) può facilmente formare radicali liberi (ad esempio, attraverso le reazioni di Fenton e Haber-Weiss). L’aumento della concentrazione di Fe 3+ è collegato al sovraccarico di FER, portando così alla ferroptosi (vedi Figura 2) [86,90].
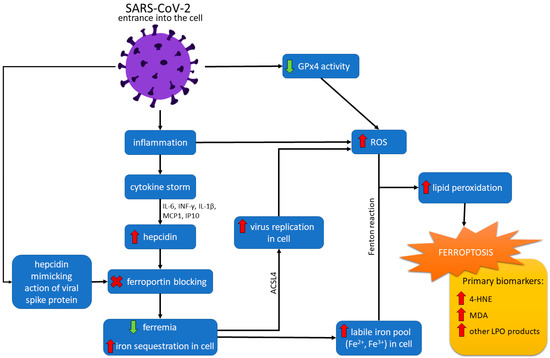
Figura 2. Vie della ferroptosi nell’infezione da SARS-CoV-2. Dopo essere entrato nella cellula, il virus SARS-CoV-2 provoca uno stato infiammatorio, con conseguente tempesta di citochine. L’effetto della tempesta di citochine, mediata da interleuchina 6 (IL-6), interferone-gamma (IFN-γ), interleuchina 1-beta (IL1-β), proteina inducibile dall’interferone 10 (IP10) e proteina chemiotattica monocitica 1 (MCP1) è un aumento del livello di epcidina. L’azione dell’epcidina sta sottoregolando la ferroportina e determinando ipoferremia e aumento del sequestro di ferro nella cellula. Il virus può anche imitare l’azione dell’epcidina mediante una proteina spike. L’aumento del livello di ferro cellulare e l’attività catalitica dell’acil-coenzima Un membro 4 della famiglia a catena lunga della sintetasi (ACSL4) promuove la replicazione del virus e aumenta la formazione di specie reattive dell’ossigeno (ROS). L’azione sottoregolata della GPx4 causata dal virus porta anche ad un aumento della produzione di ROS. Inoltre, un’elevata concentrazione di ferro cellulare può formare radicali liberi attraverso le reazioni di Fenton e Haber-Weiss. I ROS reagendo con i lipidi aumentano la perossidazione lipidica (LPO), che alla fine causa la ferroptosi.
L’infezione da SARS-CoV-2 causa alti livelli di IFN-γ, interleuchina 1-beta (IL1-β), proteina 10 inducibile dall’interferone (IP10) e proteina 1 chemiotattica dei monociti (MCP1) [87]. Pertanto, la stimolazione di queste citochine può causare la sintesi di epcidina con conseguente accumulo di ferro nei macrofagi [87].
Inoltre, l’infezione porta ad uno stato infiammatorio in cui IL-6 stimola la sintesi di FER ed epcidina [91,92]. Il virus SARS-CoV-2 induce sovraccarico di ferro promuovendo la sovraespressione di epcidina [91,92]. La possibile omologia e le connessioni evolutive tra la proteina spike del virus e l’epcidina sono oggetto di ricerca ma rimangono poco chiare [92].
SARS-CoV-2 può probabilmente imitare l’azione dell’epcidina, aumentando la FER circolante e tissutale. Ciò induce carenza di ferro sierico e abbassamento dell’emoglobina [86,89]. È stato osservato che l’aumento dei livelli sierici di epcidina e FER è effettivamente associato alla gravità dell’infezione da SARS-CoV-2 [92].
Olu et al. [93] hanno osservato un aumento dei livelli sierici di FER nei pazienti critici con COVID-19. Una ferritinemia eccessivamente alta attiva la disfunzione mitocondriale, portando a stress ossidativo mediato dal ferro e ad un peggioramento del rilascio di citochine proinfiammatorie [94]. Inoltre, livelli elevati di FER attivano il coattivatore del recettore nucleare 4 (NCOA4), mediando la ferritinofagia. La sovraespressione di NCOA4 migliora la degradazione del FER e porta al rilascio di ferro. Il ferro converte il LOOH in radicali idrossilici attraverso la reazione di Fenton, inducendo infine la ferroptosi e promuovendo il danno cellulare [95].
Il recettore della transferrina-1 (TfR-1) può facilitare l’ingresso del ferro trattenuto dalla transferrina nelle cellule [87]. TfR-1 è un portale ideale per l’ingresso di vari microrganismi nelle cellule [88]. È stato osservato che la concentrazione della proteina TfR-1 nei polmoni è significativamente esaltata nell’infezione virale [87]. Il virus può interagire proprio con questo recettore utilizzando la sua proteina spike [94].
La morte cellulare ferroptotica è una strategia di difesa contro le infezioni virali [12]. Tuttavia, prove crescenti mostrano che la morte cellulare promuove il rilascio e la diffusione del virus [96]. Kung et al. [96] hanno studiato il ruolo dell’acil-coenzima A sintetasi ACSL4 nella replicazione virale. Hanno indicato che l’attività catalitica di ACSL4 è cruciale per la replicazione virale e la ferroptosi indotta da virus. Wang et al. [97] hanno osservato una maggiore espressione di ACSL4 nei tessuti placentari infetti da SARS-CoV-2.
I ricercatori suggeriscono che il virus recluti ACSL4 per la formazione di ROS e la promozione di LPO per la replicazione virale. Di conseguenza, l’eccessiva LPO, la deplezione di GSH e un livello ridotto di GPx4 hanno portato alla morte delle cellule ferroptotiche e al rilascio del virus. È stato anche notato che la ferroptosi e gli inibitori dell’ACSL4 inibiscono significativamente la replicazione virale [96]. Wang et al. [97] affermano che ACSL4 è un buon marcatore di ferroptosi perché attiva preferenzialmente i PUFA a catena lunga per la biosintesi dei fosfolipidi e alimenta il processo ferroptotico.
È stato anche considerato il determinante principale per dettare la sensibilità alla ferroptosi e rimodellare la composizione lipidica cellulare [97]. Il virus sopprime l’espressione dell’mRNA della GPx4 associata alla ferroptosi, del TrxR correlato alla sintesi del DNA e delle selenoproteine del reticolo endoplasmatico [95]. A causa della mancanza di GPx4, il GSH non può essere perossidato per ridurre i ROS dei lipidi formati nella reazione di Fenton. Pertanto, l’accumulo di ROS causa LPO e ferroptosi (vedi Figura 2) [90].
L’infiammazione, lo stress ossidativo e l’omeostasi alterata del ferro si fondono a livello sistemico e svolgono un ruolo vitale nella patogenesi e nella progressione del COVID-19. Una conseguenza primaria della ferroptosi ferro-dipendente nell’infezione da SARS-CoV-2 potrebbe includere il deterioramento cognitivo e la perdita del gusto e dell’olfatto. Inoltre, la disfunzione mitocondriale piastrinica associata al ferro nel sangue è associata a ipercoagulopatia, che si nota generalmente nei pazienti con COVID-19 [94].
SARS-CoV-2 attacca principalmente il sistema respiratorio, ma il virus causa anche disfunzioni di altri organi, come fegato, reni o cuore [12]. Jacobs et al. [98] hanno riportato l’accumulo di fosfolipidi ossidati nel tessuto miocardico e renale nell’infezione da COVID-19.
Ciò indica che la ferroptosi contribuisce ad alcune forme di danno da ischemia-riperfusione ed è un fattore negativo nel danno cardiaco COVID-19 e nell’insufficienza multiorgano. Inoltre, il cervello dei pazienti COVID-19 sembra suscettibile alla ferroptosi [99].
Fattori essenziali della ferroptosi sono il ferro e gli acidi grassi insaturi. Il ferro è il metallo traccia più abbondante nel cervello. È necessario per il corretto metabolismo cellulare. Il cervello è sensibile allo stress ossidativo e alla LPO a causa dei suoi alti livelli di PUFA. Le membrane cellulari neuronali sono ricche di PUFA e colesterolo e suscettibili all’ossidazione mediata dai ROS innescata dalla tempesta di citochine nell’infezione da SARS-CoV-2 [99]. Skesters et al. [100] hanno osservato livelli più elevati di indicatori di stress ossidativo (MDA, 4-HNE), che sono marcatori primari di ferroptosi [99].
Ferroptosis come obiettivo terapeutico in COVID-19
La pandemia di COVID-19 è diventata una sfida considerevole e un problema di salute pubblica. Sulla base dei meccanismi sopra descritti, è ragionevole concludere che esiste un legame tra ferroptosi e COVID-19. Poiché la morte delle cellule ferroptotiche può essere coinvolta nella patogenesi di COVID-19, comprendere l’inizio di questo processo così come i suoi meccanismi regolatori sottostanti ha un potenziale significato terapeutico.
Di conseguenza, la necessità di strategie terapeutiche efficaci contro il COVID-19 è diventata una questione urgente. L’inibizione della ferroptosi può fornire metodi affidabili per il trattamento del COVID-19 [91,101]. Gli obiettivi regolatori cruciali della ferroptosi includono l’attività del sistema Xc- , il pool di ferro labile intracellulare, l’attività GPx4, la produzione di GSH, LPO e la biosintesi della fosfatidiletanolammina (PE) [99]. Il sistema Xc− , coinvolto nella sintesi del GSH, fa parte del sistema Xc-GSH-GPx4 responsabile dell’eliminazione degli effetti LPO [102]. Restrizione della Xc − sistema riduce l’assorbimento di cisteina, con conseguente carenza di GSH e la deposizione di LOOH. È potenziale, ma non ancora verificato, che la ferroptosi possa essere significativamente ridotta mediando la GPx4 con l’integrazione di selenio [102]. Si suggerisce inoltre che ACSL4 potrebbe essere un bersaglio per combattere le infezioni virali e che l’uso di inibitori ACSL4, come rosiglitazone e pioglitazone, può ridurre la carica virale dei coronavirus [96].
Inoltre, è stato proposto che i farmaci che potenziano l’asse GPx4-GSH e alla fine portano alla deplezione di ferro nel pool instabile possano essere candidati per il trattamento COVID-19 in risposta alla manifestazione della ferroptosi [103]. Pertanto, l’integrazione di GSH può essere una terapia aggiuntiva benefica in COVID-19 [85].
Inoltre, i chelanti del ferro e gli antiossidanti lipofili possono essere utili terapie aggiuntive nel trattamento del COVID-19 [12,103]. I chelanti del ferro riducono l’infiammazione e impediscono ai coronavirus di legarsi ai recettori che usano per entrare nelle cellule ospiti [103]. Edeas et al. [83] suggeriscono che i chelanti del ferro approvati, gli inibitori della ferroptosi, l’epcidina e i modulatori dell’eritropoietina possono essere presi in considerazione in aggiunta al trattamento dell’infiammazione.
Il collegamento delle citochine antinfiammatorie e l’interferenza con il metabolismo degli ioni di ferro per migliorare l’immunità dei pazienti dovrebbero essere esplorati in studi futuri [12]. Le terapie del metabolismo del ferro possono fornire benefici come potenziali farmaci per inibire l’esacerbazione dell’infezione virale causata dalla morte cellulare.
Alcuni antiossidanti, come l’idrossitoluene butilato o la vitamina E, sono riconosciuti sia come modulatori della ferroptosi che come supporto per combattere l’infezione da COVID-19 [104]. L’intervento farmacologico del corso di ferroptosi dimostra promettenti terapie per la prevenzione e il controllo delle infezioni da virus. Tuttavia, l’esatto meccanismo antivirale richiede ulteriori analisi per fornire dati di ricerca essenziali. I risultati preliminari della ricerca suggeriscono che la terapia con vitamina K potrebbe essere promettente grazie alle sue proprietà antiossidanti.
Una breve panoramica della vitamina K
Il concetto di vitamina K, scoperto nel 1929, si riferisce a composti che sono derivati di una provitamina ottenuta sinteticamente chiamata 2-metil-1,4-naftochinone (menadione, K 3 ) . La principale fonte di vitamina K per l’uomo sono le piante che la sintetizzano sotto forma di fillochinone, fitomenadione o fitonadione, cioè vitamina K 1 .
Questa forma di vitamina ha una catena laterale in posizione 3 costituita da quattro residui isoprenilici, gli ultimi tre dei quali sono saturi. Una catena con una tale struttura è chiamata residuo fitile [105,106,107,108,109,110]. La vitamina K 2 è costituita da un gruppo di derivati contenenti una catena di residui isoprenilici insaturi nell’anello naftochinonico in posizione 3. Questi composti sono menachinoni (MK). Finora sono stati descritti dodici diversi caratteri MK (da MK-4 a MK-15). Nell’uomo esiste solo MK-4 a catena corta, che è un prodotto della conversione sistemica della vitamina K1 , e quattro forme di MK-7 in MK-10, prodotti della sintesi di batteri [105,106,107,108,109,110,111,112]. Le strutture chimiche delle forme conosciute di vitamina K sono mostrate nella Figura 3.
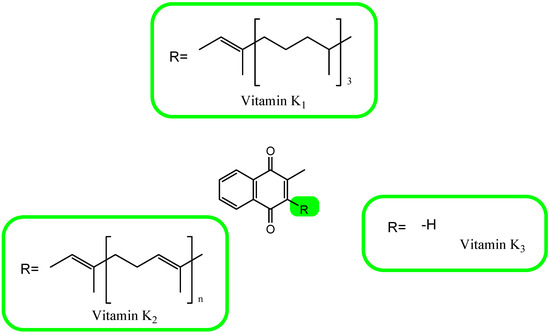
Figura 3. La formula chimica della vitamina K.
Le principali fonti di vitamina K1 sono le piante verdi e gli oli vegetali [106,110,113,114]. Carne, formaggio e prodotti a base di soia fermentata contengono una forma di vitamina K2 [ 106,108,110,114]. Poiché la vitamina K è lipofila, dopo essere stata incorporata nei chilomicroni insieme agli acidi biliari in questa forma ω, entra nel fegato. Il metabolismo della vitamina K negli epatociti inizia con la ω-idrossilazione da parte del citocromo P450 4F2, seguita dall’accorciamento della catena del poliisoprene e quindi dalla β-ossidazione ad acidi carbossilici nella posizione C5, C7 o C10 della catena laterale del poliisoprene. I metaboliti risultanti subiscono la glucuronidazione nei mitocondri e vengono quindi rimossi dal corpo sotto forma di urina e bile [110,115,116].
La funzione della vitamina K è la trasformazione post-ribosomiale delle proteine dipendenti da questa vitamina. In questa trasformazione proteica, la vitamina K funge da cofattore e i residui di acido glutammico carbossilato delle catene polipeptidiche delle proteine prodotte sono chiamati domini Gla. Le proteine con questo dominio sono conosciute come proteine Gla.
Le proteine attivate in questo processo includono i fattori della coagulazione II, VII, IX e X [107,114]. Le proteine dipendenti dalla vitamina K includono anche la proteina C, il cui compito è prevenire la coagulazione del sangue [107,117,118]; la proteina S, che funge da cofattore per la proteina C attivata e partecipa all’inattivazione dei fattori Va e VIIIa del processo di coagulazione [107,118,119]; e la proteina Z, che appartiene anch’essa ai fattori della cascata della coagulazione del sangue ed è coinvolta nella degradazione del fattore Xa [107,118,120].
Le proteine Gla si trovano in tutto il corpo e sono un componente delle ossa come l’osteocalcina, la proteina Gla della matrice (MGP) dello scheletro, la proteina Gla del rene, la proteina 6 specifica per l’arresto della crescita che è coinvolta nella stimolazione della proliferazione cellulare e la proteina ricca di Gla proteina coinvolta nella mineralizzazione dei tessuti molli [106,108,121,122].
La vitamina K è coinvolta nel processo di prevenzione dell’osteoporosi. L’interleuchina 6 (IL-6), che è un indicatore di infiammazione, influenza anche il riassorbimento osseo nel processo di osteoclastogenesi [123,124]. È stato osservato che la vitamina K riduce la produzione di IL-6 nelle colture di fibroblasti umani [125].
Uno dei ruoli importanti della vitamina K è l’impatto sulle funzioni degli osteoblasti, cioè la proliferazione, la differenziazione e l’inibizione dell’apoptosi. L’incorporazione di materia organica e minerale nella matrice ossea dipende anche dal livello di fosfatasi alcalina. Gli studi hanno dimostrato che la vitamina K aumenta l’attività della fosfatasi alcalina. In termini di osteoprotezione, la vitamina K partecipa all’attivazione del recettore degli steroidi e degli xenobiotici (SRX) e agisce come regolatore della trascrizione dei geni marcatori degli osteoblasti e dei geni della matrice extracellulare [109,126].
La carenza di vitamina K aumenta il rischio di infiammazioni croniche [112]. La ferroptosi è stata al centro dell’attenzione nella ricerca di metodi di trattamento del cancro. Esiste un legame tra la ferroptosi e la vitamina K. Uno degli effetti della ferroptosi, come descritto nella Sezione 3, è la formazione di prodotti di perossidazione lipidica. La vitamina K può essere trovata in tre forme nel corpo. Queste sono rispettivamente la forma ridotta (idrochinone), la forma ossidata (chinone) e la forma epossido della vitamina K [109].
La forma ridotta della vitamina K inibisce la perossidazione lipidica trattenendo i radicali liberi nella membrana plasmatica. La vitamina K sfrutta l’attività della proteina 1 soppressore della ferroptosi (FSP1), con attività reduttasica a scapito del NAD(P)H, e grazie ad essa mantiene alto il livello della forma ridotta. Questo fa sì che la vitamina K abbia proprietà antiossidanti definite [127,128]. In uno studio significativo, Mishima et al. [127] hanno appena dimostrato che FSP1 mantiene alti livelli di vitamina K ridotta, sopprimendo così la ferroptosi.
Nei loro studi sulle linee cellulari, i ricercatori indicano che FSP1 è un regolatore cruciale della ferroptosi che opera separatamente da GPX4. Questa ubichinone ossidoreduttasi NAD(P)H-dipendente svolge un ruolo significativo nella ferroptosi convertendo l’ubichinone in ubichinolo e utilizzando NAD(P)H [127]. Conclusioni simili sono state raggiunte da Kolbrink et al. [129], che ha dimostrato che la vitamina K 1 è un forte inibitore della ferroptosi e può essere usata come medicinale nel corso di danno renale acuto.
È stato anche dimostrato che la vitamina K 2 inibisce la ferroptosi e protegge dalla morte cellulare causata dallo stress ossidativo. Questo effetto sembra essere mediato almeno in parte dall’attivazione della via antiossidante Nrf2. Il percorso Nrf2 è un meccanismo di difesa cellulare che aiuta a proteggere le cellule dallo stress ossidativo sovraregolando l’espressione dei geni coinvolti nella difesa antiossidante e nella disintossicazione.
È stato dimostrato che la vitamina K2 attiva questa via, portando ad una maggiore espressione di enzimi antiossidanti, tra cui la glutatione perossidasi [130,131,132]. Dato il ruolo potenziale della ferroptosi nella patogenesi del COVID-19 e il possibile legame tra vitamina K e ferroptosi, c’è interesse nell’esplorare i potenziali benefici terapeutici della vitamina K nel COVID-19. Tuttavia, sono necessarie ulteriori ricerche per comprendere appieno i meccanismi alla base della relazione tra vitamina K, ferroptosi e COVID-19 e per determinare se l’integrazione di vitamina K potrebbe essere una strategia terapeutica sicura ed efficace per COVID-19.
Vitamina K nel COVID-19
Lo scoppio della pandemia causata da SARS-CoV-2 ha spinto gli scienziati a cercare composti biologicamente attivi che abbiano effetti antivirali e immunomodulatori. Nei pazienti con SARS-CoV-2, lo stress ossidativo è associato all’amplificazione e al mantenimento della tempesta di citochine e della coagulopatia [3].
Il COVID-19 induce uno stato di ipercoagulazione che spesso porta a complicanze tromboemboliche [133,134]. Nei pazienti ospedalizzati è stata osservata un’accelerazione della fibra elastica dovuta a mineralizzazione e degradazione [135]. Questo è il motivo per cui la vitamina K, che influenza l’attivazione dei fattori anti- e pro-coagulazione rispettivamente nei tessuti periferici e nel fegato, era di interesse [136,137,138].
La vitamina K partecipa alla protezione dei polmoni contro la calcificazione e il danno [137,139,140]. Ciò che viene utilizzato per l’identificazione dello stato extraepatico della vitamina K è la proteina Gla della matrice defosforilata non carbossilata (dp-ucMGP) e il rapporto tra osteocalcina non carbossilata e carbossilata [135,137,141,142].
Gli alti livelli di dp-ucMGP riflettono uno stato basso di vitamina K e viceversa [142]. Carenze di vitamina K sono state dimostrate negli adulti ospedalizzati con infezione da COVID-19 [135,138,141,143]. Un basso stato di vitamina K prevede una mortalità più elevata tra i pazienti con COVID-19 [138].
Poiché la vitamina K ha attività antinfiammatoria e offre protezione contro lo stress ossidativo, influenza il decorso della fase iniziale dell’infezione acuta da COVID-19 [141,143,144]. MGP, attivato dalla vitamina K, inibisce la degradazione delle fibre elastiche e la mineralizzazione vascolare [135].
L’aumento dell’uso di vitamina K per la carbossilazione dell’MGP polmonare e i fattori della coagulazione influenzano il decorso della malattia [135,138]. Non sono i bassi livelli basali di vitamina K a essere responsabili della carenza extraepatica di vitamina K, ma principalmente l’aumento dell’uso di vitamina K durante l’infezione [142].
Gli effetti dei derivati della vitamina K sulle cellule immunitarie umane non sono stati ampiamente studiati. In un modello animale, è stato dimostrato che il menadione può essere un’efficace strategia terapeutica contro il danno polmonare acuto, inclusa l’ARDS. La vitamina K3 inibisce l’attivazione di NF-κB, che è necessaria per l’espressione delle citochine e le seguenti risposte infiammatorie nel modello di topo ARDS (la linea cellulare RAW264.7 simile ai macrofagi di topo) [145].
I livelli di derivati della vitamina K sono inversamente correlati con i livelli di citochine infiammatorie, tra cui IL-6, fattore di necrosi tumorale α (TNF-α) e proteina C-reattiva (CRP) [124,146]. Possono aumentare la frequenza delle cellule CD4 + CD25 + Foxp3 + T regolatorie (Treg), che svolgono un ruolo vitale nel mantenimento dell’omeostasi immunitaria e nella prevenzione delle risposte autoimmuni [147].
Sebbene la carenza di vitamina K sia costantemente associata a esiti clinici peggiori nei pazienti COVID-19, non è stato dimostrato in modo definitivo che l’integrazione di vitamina K abbia il potenziale per prevenire o migliorare gli esiti aumentando l’attivazione delle MGP polmonari e della proteina S endoteliale [135].
I livelli circolanti di vitamina K geneticamente predetti studiati non forniscono alcuna prova che l’integrazione di vitamina K possa prevenire l’infezione da SARS-CoV-2 o il ricovero dovuto a COVID-19 [136]. L’uso regolare di antagonisti della vitamina K (AVK) come farmaci anticoagulanti riduce drasticamente la biodisponibilità della vitamina K attiva.
L’uso di VKA prima di COVID-19 è associato ad un aumentato rischio di mortalità nei pazienti COVID-19 [148]. La carenza di vitamina K è associata a fattori che aumentano il rischio di COVID-19 grave e fatale, inclusi dati demografici, indice di massa corporea (BMI), marcatori infiammatori e comorbidità (malattie cardiovascolari, polmonari e renali) [138,139,141,142].
I ruoli protettivi della vitamina K nell’infiammazione possono ridurre l’effetto delle sequele immunitarie avverse nel caso dell’infezione da SARS-CoV-2. Sono necessarie ulteriori ricerche sull’importanza dell’integrazione di vitamina K nella prevenzione e nel trattamento del COVID-19 grave.



{kind=link}