











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



There is no cure for Alzheimer’s, and the available treatments only offer symptomatic relief.
However, a new drug called Leqembi (lecanemab-irmb – Synonyms: Lecanemab-irmb, BAN2401, mAb158) may change that.
BACKGROUND
BAN2401 is the humanized IgG1 version of the mouse monoclonal antibody mAb158, which selectively binds to large, soluble Aβ protofibrils. This therapeutic antibody grew out of the discovery of the “Arctic” mutation in APP, which leads to a form of clinically typical Alzheimer’s disease that is marked by particularly high levels of Aβ protofibrils and relative absence of amyloid plaques (see Nilsberth et al., 2001). mAb158 was originally developed at Uppsala University, Sweden (Englund et al., 2007).
In its preclinical development, mAb158 was found to reduce Aβ protofibrils in brain and CSF of Tg-ArcSwe mice (Lord et al., 2009, Tucker et al., 2015). Subsequent studies in mouse neuron-glial co-cultures showed that mAb158 may protect neurons, i.e., reduce Aβ protofibril toxicity, by counteracting the pathological accumulation of these protofibrils in astrocytes (Söllvander et al., 2018). In a direct comparison with the anti-amyloid antibodies aducanumab and gantenerumab, lecanemab was reported to bind most strongly to Aβ protofibrils, while the others preferred more highly aggregated forms (Nov 2021 conference news). Lecanemab was shown to bind to diffusible Aβ fibrils from human brain tissue (Nov 2022 news).
BAN2401 was licensed to Eisai, which in March 2014 signed a collaboration agreement with Biogen for joint development of this therapeutic antibody.
While clinical trials are being conducted in Alzheimer’s (see below), preclinical research with postmortem Down’s syndrome brain sections indicates binding of lecanemab to Aβ deposits in this disease, as well (Johannesson et al., 2021).
FINDINGS
A multicenter Phase 1 trial tested the safety, tolerability, and pharmacokinetics of single and multiple ascending intravenous doses of BAN2401 in 80 people with mild to moderate AD. Changes in Aβ levels were also measured. BAN2401 was well-tolerated at all doses tested, up to 10mg/kg every two weeks for four months, with amyloid-related imaging abnormalities (ARIA-E, ARIA-H) occurring at the same rate in both placebo and BAN2401. The antibody entered the CSF and showed dose-dependent exposure, though with a short serum elimination half-life of seven days and no clear effect on CSF biomarkers. Results were published (Logovinsky et al., 2016).
Subsequently, a Phase 2, 18-month U.S. trial tested five different intravenous doses of BAN2401 in a Bayesian adaptive design. Allocation of subsequent enrollees to different groups was adjusted in response to frequent interim analyses, the first to be done in late 2015 after the first 196 patients had entered the trial, and again every time 50 more people had enrolled (for detailed description of this innovative trial design see Satlin et al., 2016). This trial enrolled 856 people who had either early stage AD as defined by the proposed NIA-AA diagnostic criteria or mild cognitive impairment due to AD, or who met NIA-AA criteria for probable AD and whose diagnosis was confirmed by a positive amyloid PET scan. As primary outcomes, the trial measured 12-month change from baseline in the new ADCOMS composite of cognitive tests (Wang et al., 2016), and safety.
In 2017 the sponsors announced that BAN2401 had shown no cognitive benefit at this 12-month time point. However, futility conditions had not been met either at the 17 interim analyses conducted until then. Therefore the trial continued to full enrollment of 856 participants, and out to the full treatment period of 18 months (Dec 2017 news). In February 2018, the trial protocol was amended to offer up to five years of additional treatment in an open-label extension phase, in which change on the ADCOMS will be measured at each visit.
The sponsors announced top-line results of the blinded 18-month treatment phase in July 2018 (see July 2018 news). The highest antibody dose of twice-monthly 10 mg/kg slowed progression on the ADCOMS and reduced brain amyloid accumulation, according to a press release from BioArctic. Full results of this Phase 2b study were presented at AAIC (Jul 2018 news). The antibody reduced brain amyloid by up to 93 percent in the highest-dose group. This dose slowed cognitive decline by 47 percent on the ADAS-Cog, and by 30 percent on the ADCOMS. The next-lower dose, 10 mg/kg monthly, showed a trend toward slower cognitive decline that was not statistically significant. In an analysis of CSF from a subgroup of patients, the treatment caused a dose-dependent rise in CSF Aβ42. MRI scans detected ARIA in just under 10 percent of participants overall, and in fewer than 15 percent of those with ApoE4 in the highest-dose group. Most ARIA occurrences were asymptomatic.
The results were complicated by uneven distribution of ApoE4 carriers between placebo and treatment groups, which was caused by an EMA request during the trial. A subgroup analysis, presented at CTAD, suggested that the treatment benefit was not due to this imbalance (Nov 2018 conference news). Full results were subsequently published (Swanson et al., 2021). Additional analysis of the three cognitive endpoints with six different statistical methods found a consistent positive effect of treatment (Nov 2021 conference news).
An open-label extension to this trial re-enrolled its participants to deliver the highest antibody dose for up to two years total. As reported at AD/PD 2019 in Lisbon, Portugal, Eisai/Biogen planned to treat up to 250 people in this extension, which was to run until August 2021 (May 2019 conference news). Baseline data from 35 participants suggested that brain amyloid load had remained steady during a two-year pause in antibody dosing, but that cognition declined when BAN2401 was discontinued (Dec 2019 conference news).
One-year brain imaging data from 76 participants, presented in December 2020 at CTAD, indicated that people previously treated with placebo had large decreases in their brain amyloid since entering the OLE, while those previously treated with antibody maintained low levels of brain amyloid. ARIA-E incidence was comparable to the core study. Most ARIA-E was asymptomatic and resolved within four to 12 weeks. Continuing to dose people with mild to moderate ARIA-E appeared to pose no additional safety issues (Nov 2020 conference news). More one-year OLE data on 180 participants was presented in March 2021 at AD/PD. Brain amyloid fell fastest in those who began the OLE with the highest amyloid. By the end, 80 percent of participants were judged amyloid-negative, with SUVRs below 1.17 (Mar 2021 conference news).
At the July 2021 AAIC, Eisai reported 18-month OLE data on 100 people. It suggested a slowing of cognitive decline in the open-label phase, compared to ADNI historical data. CSF Aβ42/40, which had increased with treatment during the placebo-controlled phase of the trial, started to decline during the dosing gap, but rose again in all participants after open-label treatment began. A post hoc analysis across the entire study duration suggested cognition declined more slowly in people on lecanemab than in those on placebo (Aug 2021 conference news). More data presented at the Nov 2021 CTAD showed a correlation between plaque clearance and slowed decline on ADCOMs in the OLE (Nov 2021 conference news). At the same conference, Eisai reported that changes in plasma ptau181 tracked with changes in amyloid PET and plasma Aβ42/40. OLE results were formally published (McDade et al., 2022), as were details on ARIA in the completed study (Honig et al., 2023), and an explication of the advantages of the Bayesian design (Berry et al., 2023).
In March 2019, Eisai began a Phase 3 trial called Clarity AD, to be run at 250 sites across the world. It aimed to enroll 1,566 people with early symptomatic AD, who received 10 mg/kg drug or placebo every two weeks for 18 months, followed by a two-year open-label extension. The primary outcome in the core study was change in CDR-SB at 18 months, with secondary outcomes of brain amyloid, ADCOMS, and ADAS-Cog14 subscale. In the extension phase, primary outcomes were change in CDR-SB as well as safety. Change in the CSF biomarkers neurogranin, neurofilament light chain, Aβ(1-42), total tau, and phospho-tau from baseline up to 45 months was originally listed as a primary outcome in the trial registration, but this was dropped in July 2019. Plasma and CSF biomarkers, as well as amyloid and tau PET, are being assessed in optional longitudinal substudies.
As of October 2020, Clarity AD had randomized 1,222 participants, with demographic and cognitive scores similar to the Phase 2 study (Nov 2020 conference news). By March 2021, it had exceeded its enrollment goal, at 1,794 patients, the company announced (press release). The trial is set to run until 2027.
In February 2020, the Alzheimer’s Therapeutic Research Institute announced that the Alzheimer’s Clinical Trial Consortium (ACTC) would conduct a large Phase 3 study of lecanemab, called AHEAD 3-45 and co-funded by the NIH and Eisai (press release). It started in July 2020 as a four-year trial that comprises two sub-studies in a combined 1,400 people who are cognitively normal but have elevated brain amyloid. A3 is enrolling 400 people whose amyloid is below the brain-wide threshold for positivity; they will receive 5 mg/kg titrating to 10 mg/kg BAN2401 or placebo every four weeks for 216 weeks, and their primary outcome will be change in brain amyloid PET at that time. A45 is enroll 1,000 participants who have a positive amyloid PET scan.
They will receive BAN2401 titrated to 10 mg/kg every two weeks for 96 weeks, followed by 10 mg/kg every four weeks through week 216. Their primary outcome is change from baseline on their Preclinical Alzheimer Cognitive Composite 5 (PACC5) score, also at week 216. Secondary outcomes for A45 include change in brain amyloid PET and cognitive function.
Both studies will measure change in tau PET as a secondary outcome. This new study will use blood Aβ42/40 measures to rule out participants unlikely to have elevated brain amyloid before proceeding to PET imaging (for details, see Rafii et al., 2022).The first person was dosed in this trial in September 2020; as of the July 2021 AAIC, 77 people had been randomized. As of November 2022, 107 sites were recruiting around the world. The trial is expected to run until October 2027.
In June 2021, the FDA designated lecanemab a breakthrough therapy, expediting regulatory review, and soon after, Eisai/Biogen began their application for marketing approval (press release, Oct 2021 news). In December 2021, the agency granted fast-track designation (press release).
In September 2021, Eisai began a Phase 1 evaluation of lecanemab subcutaneous administration. The study compared the pharmacokinetics, bioavailability, and safety of a single 700 mg injection under the skin in the abdomen to 10 mg/kg given intravenously in 60 healthy people; it was completed in January 2022. In September 2022, a Phase 1 study started evaluating bioequivalence of a subcutaneous formulation delivered with an auto-injector device; it aims to enroll 160 healthy participants and be finished by February 2023.
In November 2021, the DIAN-TU announced its choice of lecanemab for the first DIAN-TU anti-amyloid/anti-tau concurrent trial (Nov 2021 conference news). It will pair lecanemab with the anti-tau antibody E2814 in the Tau NexGen prevention study in 168 people with familial AD mutations. All will receive lecanemab, while half will get E2814, and half a matching placebo, against a primary outcome of change in tau PET. This trial will run at 40 sites in the Americas, Australia, Japan, and six European countries, until 2027 (Mar 2021 news).
In May 2022, Eisai/Biogen completed lecanemab’s FDA submission, based on the Phase 2 data (May 2022 news). In July 2022, the FDA granted priority review status to the application, with a decision date of January 6, 2023 (Jul 2022 news).
On September 27, 2022, Biogen and Eisai reported positive top-line results on all primary and secondary outcomes of the Phase 3 Clarity AD study (Sep 2022 news). Results were presented in December at the 2022 CTAD conference, and published the same day (Dec 2022 conference news; van Dyck et al., 2022). Lecanemab slowed decline on the CDR-SB by 27 percent compared to placebo at 18 months. Key secondary endpoints, including the ADAS-Cog14, ADCOMS, and ADCS MCI-ADL, all showed a similar slowing of decline. Two-thirds of the treated group became PET-amyloid negative at 18 months.
Tau PET indicated a significant slowing of tangle accumulation in the medial temporal lobe, and trended toward slowing in other brain regions. The astrocytosis biomarker GFAP was reduced on treatment, but markers of neurodegeneration were mixed. Safety was in line with past studies, with amyloid-related ARIA-E detected in 12.6 percent of treated participants vs 1.7 percent of the placebo group. About one-quarter had symptoms, which were typically mild and transient. Three people had severe symptoms, but the company did not say what they were.
Three deaths from brain hemorrhage have been reported in the lecanemab open-label extension (Jan 2023 news). Two of the three people had received blood thinners. In Clarity, macrohemorrhages, defined as any brain bleed larger than 1 cm, occurred in 0.6 percent in the treatment group, and 0.1 percent in the placebo group. For people on anticoagulants and lecanemab, the rate increased to 2.4 percent. According to a news article, the third fatality was a woman with preexisting cerebral amyloid angiopathy (Piller 2023).
On January 6, 2023, the FDA approved lecanemab under the Accelerated Approval pathway, based on evidence of effect on the surrogate endpoint of amyloid removal in the Phase 2 trial, and a reasonable likelihood of clinical benefit (FDA press release; Jan 2023 news). Lecanemab will be sold under the brand name Leqembi.
In March 2023 , the U.S. Veterans Health Administration announced it would cover the cost of lecanemab for veterans in the early stages of AD, excluding those with two copies of APOE4 (press release; VA use criteria).
By July 6, the FDA will decide whether to grant a traditional, full approval, after considering Phase 3 data (Mar 2023 news).
Additional efficacy data from the Clarity trial, presented at the March 2023 AD/PD conference, showed benefit on self-reported outcomes capturing quality of life and caregiver burden (April 2023 news).
In March 2023, Leqembi Appropriate Use Recommendations were published (Cummings et al., 2023). They stipulate that it not be prescribed for people taking anticoagulants, or those with a clotting disorder, strokes, or seizures. People on lecanemab should not be treated with acute thrombolytics such as tPA, though common antiplatelet medications such as aspirin or clopidogrel are allowed. The AUR recommend APOE genotyping prior to therapy, so clinicians can discuss risks with patients, but allow treatment for APOE4 homozygotes. A schedule of monitoring MRIs is recommended to include scans at baseline, and before the fifth, seventh, and 14th biweekly infusions. Scans are recommended at one year for APOE4 carriers and people with any ARIA during the first year.
“For all clinical trials of Leqembi/lecanemab, see clinicaltrials.gov.”
Social Impact of AD in the U.S.
According to the Alzheimer’s Association’s “2022 Alzheimer’s Disease Facts and Figures”1, an estimated 6.5 million Americans aged 65 and older are living with dementia due to AD (i.e., mild, moderate and severe dementia stages of AD). AD was officially listed as the sixth-leading cause of death in the U.S. in 2019 and the seventh-leading cause of death in 2020 and 2021. It is a chronic, progressive, disabling and fatal disease. According to another report from the Alzheimer’s Association, “Changing the Trajectory of Alzheimer’s Disease: How a Treatment by 2025 Saves Lives and Dollars”2, the total costs of care in the U.S. from all payers (i.e., Medicare, Medicaid, out-of-pocket and other payers), would increase from $267 Billion in 2020 to $451 Billion in 2030 if no treatment exists to delay the disease.
A recent 2022 study, “Projecting the Long-term Societal Values of Disease-Modifying Treatment for Alzheimer’s Disease in the United States”3, quoted as many as 10 to 14 million Americans living with MCI (Mild Cognitive Impairment) of any etiology, the very mild symptomatic stage before onset of dementia stages, in which 55% have AD as the underlying pathology. In this study, the lifetime value of a disease-modifying treatment in Early AD (MCI due to AD and mild dementia stage of AD) from a U.S. societal perspective assuming a treatment effect of 30% relative decline in progression rates from MCI to mild dementia and from mild to moderate dementia was estimated at $134,418 per person.
Value of LEQEMBI Adoption to U.S. Society
LEQEMBI is indicated for the treatment of AD, and treatment with LEQEMBI should be initiated in patients with MCI or mild dementia stage of AD after confirmation of amyloid beta pathology (Early AD). In the U.S., we estimate that the diagnosed eligible Early AD population will reach approximately 100,000 individuals by year 3 representing a measured initial attainment in the real world and will increase gradually over the mid-to-long term given the time required to advance new screening and diagnostic technologies such as blood-based biomarkers to confirm amyloid beta pathology.
Published findings in a peer-reviewed journal4 from the confirmatory Phase 3 Clarity AD study in patients with Early AD demonstrate that LEQEMBI treatment resulted in less decline on measures of cognition and function than placebo at 18 months (27% slowing over 18 months measured by CDR-SB*) and was associated with adverse events that were within expectation. Clarity AD results were consistent with that from the Phase 2 trial (Study 201)5 that was the basis for the FDA’s accelerated approval and will be submitted to the FDA very shortly for review for traditional approval.
LEQEMBI’s clinical data show that it could help patients maintain cognition, preserve activities of daily living and maintain functional ability for longer, and therefore, LEQEMBI’s clinical efficacy could potentially translate into impactful outcomes for these patients and their families. A simulation study using an established, validated disease model called Alzheimer’s disease Archimedes condition event (AD ACE),6,7,8 which is an individual patient-level model with a focus on predicting the trajectory of cognitive decline and simulating the effects of early interventions in AD, helped assess the potential lifetime value and economic impact of LEQEMBI in patients with Early AD based on clinical trial populations and findings. The results9,10 of this simulation study based on Study 201 of LEQEMBI were published in a peer-reviewed journal, and the model has been recently updated with data from the Clarity AD trial showing consistent outputs.
Per Patient Societal Value of LEQEMBI at $37,600 per Year in the U.S. by AD ACE Model
In the updated AD ACE simulation study using Clarity AD data, LEQEMBI treatment was projected to delay disease progression, resulting in an increase in patient’s expected time in Early AD while reducing the time in more advanced severe states. Slowing of clinical decline in patients treated with LEQEMBI is estimated to delay disease progression by nearly 3 years on average (delay progression mainly from MCI to mild AD and from mild to moderate AD) compared to standard-of-care (SOC).
LEQEMBI’s impact on the disease trajectory is then modeled into an annual per-patient value to the U.S. society based on four components according to the following equation: a) quality-adjusted life-years (QALY) gains compared to SOC; b) willingness-to-pay (WTP) threshold; c) cost offsets compared to SOC; and d) time on treatment, all in present value term. Note that the costs, benefits and time on treatment need to be adjusted for the values between present time and future times using discounting in a health economic evaluation.
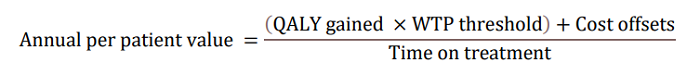
With regard to a) QALY gains, since health is a function of length of life and quality of life, this measure combines both attributes into one single index, with one QALY gain representing one additional year of a person’s life at perfect health. LEQEMBI treatment was predicted to offer an additional 0.64 QALYs compared to SOC for an Early AD patient over lifetime by improving outcomes for both the patient and caregiver. With regard to b) WTP threshold per QALY gained, conventionally, the WTP threshold is based on 1 to 3 times country’s per capita gross domestic product (GDP). In the U.S., as a result, a WTP threshold of $50,000 to $150,000 is referenced as the cost-effectiveness threshold11, but a higher WTP threshold is often considered to account for the gravity of conditions with greater burden, and interventions that exhibit wider societal benefits, such as AD with substantial impacts on caregivers.
A modified societal perspective at $200,000 WTP threshold per QALY gained is used in the U.S. when societal cost of the disease is large such as AD. With regard to c) cost offsets, avoidance of both direct medical/non-medical costs for AD management, as well as indirect costs of caregivers including family members totaling $7,415 over the lifetime for each Early AD patient, was predicted with LEQEMBI treatment compared to SOC.
Direct costs contain cost of medications, medical visits, hospitalizations, living accommodations and community services for the patients. Indirect costs of caregivers, including family members, consider the monetary value for hours spent on caregiving activities. With regard to: d) time on treatment, LEQEMBI was modeled to be stopped upon transition to moderate AD dementia or worse. The average treatment duration in this Early AD population was estimated to be approximately 3.6 years with consideration of discounting to present value.
Taking all four components together in present value term, for a) 0.64 QALYs gained b) at WTP threshold of $200,000 per QALY gained plus c) cost offsets of $7,415 over d) 3.6 years on treatment, the yearly per-patient value of LEQEMBI from a societal perspective was quantified at approximately $37,600.
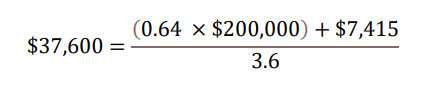
This yearly estimate of $37,600 equals to approximately $135,000 (0.64 × $200,000 + $7,415) in lifetime value per patient to the U.S. society. Over 10 years cumulatively, the gradual adoption of LEQEMBI treatment among Early AD patients could potentially generate positive social impact of several tens of billion dollars to the U.S. society, in the form of “clinical value” that help patients delay their disease progression, projected “social value” that help improve patients’ and caregivers’ quality of life and productivity, and simulated “economic value” that help reduce demand for health services.
LEQEMBI U.S. Launch Pricing at $26,500 per Year
While we estimate the per-patient-per-year value of LEQEMBI treatment to the U.S. society to be $37,600, Eisai decided to price LEQEMBI below quantified societal value at the wholesale acquisition cost (WAC) of $26,500 per year (estimated annual price based on 10mg/kg IV biweekly for average U.S. patient weight of 75kg based on Study 201 and Clarity AD) aiming to promote broader patient access, reduce overall financial burden, and support health system sustainability. As such, the WAC for the 200mg vial is $254.81 and the WAC for the 500mg vial is $637.02.
Actual annualized pricing may vary by patient. In addition, Eisai continues to pursue a less frequent maintenance dosing regimen for LEQEMBI, such as monthly instead of current biweekly regimen, upon significant amyloid beta clearance to prevent re-accumulation of amyloid beta biomarkers while maintaining clinical efficacy. This could further lower the yearly cost of LEQEMBI during the maintenance dosing phase, for example, from $26,500 to potentially about half of this figure given less amount of drugs.
Patient Affordability
Eisai believes patient affordability must be a key consideration to promote patient access and intended use and benefits of LEQEMBI. Among the eligible Early AD patient population in the U.S., once the patient’s insurer covers LEQEMBI, we estimate that approximately 91% of individuals will be covered by Medicare with Medigap (supplemental insurance), Medicare Advantage (Medicare-approved plans from private companies with potential supplemental coverage), Medicaid, and Commercial (private insurance12).
For these patients, their out-of-pocket costs for LEQEMBI treatment could range from $0 to a few dollars per day. Remaining 9% of the individuals will fall into the category of Medicare without supplemental insurance, and hence will be responsible for 20% of the LEQEMBI cost as co-insurance under Medicare Part B. For these patients, their estimated out-of-pocket costs for LEQEMBI at the price of $26,500 per year will translate into about $14.50 per day13. Across the entire eligible Early AD patient population, we estimate the weighted average out-of-pocket costs for LEQEMBI to be about $2 per day12.
Commitment to Patient Access
Eisai is committed to ensuring that certain financially disadvantaged patients have access to LEQEMBI. Firstly, Eisai is establishing a Patient Assistance Program, which will provide LEQEMBI at no cost, for eligible uninsured and underinsured patients, including Medicare beneficiaries, who meet financial need and other program criteria. Secondly, Eisai will offer patient support for improving access through LEQEMBI Patient Navigators, who will provide information about accessing LEQEMBI, help patients and their families understand their insurance coverage and options, and identify financial support programs for eligible patients.
Health System Sustainability
We believe our pricing approach for LEQEMBI would also help improve health system sustainability, which is projected based on appropriate use of LEQEMBI in eligible patients with Early AD to improve patient’s health outcomes and quality of life and reduce demand for health services and global burden of disease through changing disease trajectory. Furthermore, we believe our pricing approach for LEQEMBI, coupled with the size of the targeted patient population, will be sustainable under historical growth and spending assumptions for Medicare Part B.
Giving Back More Than Half of LEQEMBI Value to U.S. Society
The price of LEQEMBI at a yearly cost of $26,500 is $11,100 below the projected societal value of $37,600, and a less frequent maintenance dosing regimen will further lower the yearly cost well below the projected societal value. Taking these savings as well as discounts and rebates within the U.S. healthcare system into consideration, over 10 years cumulatively, the gradual adoption of LEQEMBI treatment at this pricing approach could give back about 60% of the potential positive social impact of several tens of billion dollars to the U.S. society14.
These resources could help realize new innovations that enhance the health and quality of life of individuals at risk of developing or living with AD as well as their families and caregivers. On the other hand, about 40% of the potential positive social impact of several tens of billion dollars will be accrued by employees and shareholders in the form of product sales, from which we are committed to re-invest in future research and development to create new AD therapies and new innovations such as establishing ecosystems toward inclusive AD communities14. We deeply believe that our pricing approach to maximize value for all stakeholders will help Eisai achieve social good in the form of relieving anxiety over health and reducing health disparity according to our corporate philosophy.
refereence link : https://www.alzforum.org/therapeutics/leqembi
clinicaltrials.gov.
https://www.eisai.com/news/2023/news202302.html



{kind=link}