











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



In studies in mice, Johns Hopkins Medicine researchers report they have found that bilirubin, a bile pigment most commonly known for yellowing the skin of people with jaundice, may play an unexpected role in protecting brain cells from damage from oxidative stress.
Bilirubin is commonly measured in lab tests as a marker for liver or blood health, and high levels may indicate disease.
However, whether it has a role in healthy people has remained unclear.
The Johns Hopkins Medicine team says its interest in the compound’s function in the brain arose from testing which tissues in the mouse body produced bilirubin.
Surprisingly, the researchers found “exceptional levels” of the stuff in mouse brains — five to 10 times higher production than in rodents’ livers.
“Bilirubin is normally considered a waste product, but this level of production takes a lot of metabolic energy, and it seemed bizarre for bilirubin to not have a function,” says Bindu Paul, Ph.D., faculty research instructor at the Johns Hopkins University School of Medicine’s Solomon H. Snyder Department of Neuroscience, and a member of the research team.
The new study, described in a report published July 25 in Cell Chemical Biology, set out to find the purpose for harboring so much bilirubin in the brain.
The team noted that past studies proposed that bilirubin might be an important antioxidant.
Since the brain is so metabolically active and vulnerable to oxidative damage, the research group considered the possibility that bilirubin might be particularly important to protecting the brain against oxidative stress.
For their experiments, the team used mouse neurons grown in the laboratory that were genetically engineered to not produce bilirubin.
As the cells grew, the researchers exposed them to various sources of oxidative stress by introducing reactive molecules to their environment.
When compared with normal mouse brain cells, the researchers found that the genetically modified mouse neurons were far more vulnerable to these stressors — particularly at the hand of a harmful form of oxygen called superoxide.
Chirag Vasavda, an M.D./Ph.D. student in Solomon Snyder’s laboratory and first author on the study, notes that superoxide is an important chemical cell messenger linked to learning, memory and development in the brain.
However, excessive brain cell activity can lead to uncontrolled superoxide levels, which can trigger oxidative stress and initiate a series of harmful reactions that cause damage to the brain.
“Our initial experiments hinted to us that bilirubin might play an important role in controlling the levels of superoxide in the brain,” says Vasavda.

Collage of neuron cells used in this study. Neurons here are expressing a protein that fluoresces upon binding bilirubin. The image is credited to Chirag Vasavda.
The research team suspected that bilirubin’s ability to regulate superoxide originated in its chemical structure, which allows it to grab on to and neutralize the harmful molecule in a way that other antioxidants, such as glutathione and cysteine, cannot.
To test this, the researchers stimulated excessive brain cell activity in normal brains and brains engineered to lack bilirubin.
They found that brains lacking the bilirubin-production gene accumulated excessive superoxide.
Then they stimulated brain activity in normal mice and mice lacking bilirubin to test whether removing bilirubin worsens brain damage or cell death.
The researchers found that mice that lacked bilirubin had about two to three times more brain damage as their normal counterparts, suggesting that bilirubin protected normal brains against harmful superoxide reactions.
This discovery, the investigators say, advances scientific understanding of bilirubin’s role in the brain and elsewhere and could lead to novel treatments for neurodegenerative diseases such as Huntington’s and Parkinson’s that are marked by excessive superoxide levels and oxidative stress.
Other researchers involved in this study include Ruchita Kothari, Adarsha Malla, Ming Ji, Cristina Ricco, Risheng Xu, Harry Saavedra, Juan Sbodio, Adele Snowman, Lauren Albacarys, Lynda Hester, Thomas Sedlak and Solomon H. Snyder of the Johns Hopkins University School of Medicine; Robert Tokhunts of Dartmouth College; and Anthony Lin of the Duke University School of Medicine.
Funding: This work was supported by the National Institute of Mental Health (MH18510), the National Institute of General Medical Sciences (T32 GM73009) and the National Institutes of Health Office of the Director (S1D016374, S1OD016374).
The authors declare no competing financial interests.
Bilirubin Production and Excretion
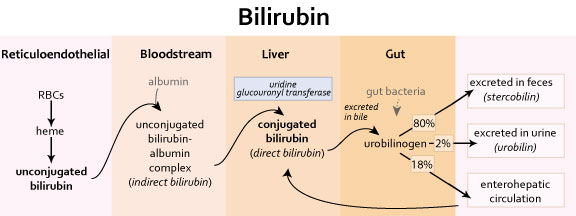
| Pathway |
| Steps from bilirubin production to excretion (see above) (1) reticuloendothelial system (RES) – macrophages phagocytose sensescent erythrocytes – hemoglobin metabolism yields bilirubin – pathway: heme → biliverdin (green-colored) → bilirubin (yellow-colored) (2) bloodstream – albumin binds bilirubin and complex is carried to liver – bilirubin-albumin complex = indirect bilirubin (water insoluble) (3) liver – hepatocytes take up bilirubin – hepatic microsomes conjugate bilirubin with glucoronic acid – conjugation via UDP glucuronyl transferase -enzyme is synthesized slowly after birth, sometimes causing newborn jaundice – conjugated bilirubin = direct bilirubin aka water soluble – a portion of conjugated bilirubin is excreted in urine – remainder is secreted into bile and then into small intestine (4) gastointestinal tract – in terminal ileum and colon, bilirubin is deconjugated by bacterial enzymes and metabolized to urobilinogen – 18% of urobilinogen is absorbed via enterohepatic circulation and delivered back to liver – 80% of urobilinogen is converted to stercobilin and excreted in feces – stercobilin gives characteristic color of feces – 2% of urobilinogen is converted to urobilin and excreted in urine – urobilin gives characteristic color of urine |
*-*-*-*-*
Alzheimer’s disease (AD) is the most common neurodegenerative disease.
Its prevalence is increasing markedly worldwide due to ageing populations and a lack of disease modifying therapies.
AD clinically manifests as memory deficits in combination with a progressive deterioration of other cognitive domains such as executive function and visuospatial skills.
At post mortem examination severely affected AD brains are commonly reduced to half their original volume1 with a disproportionate neuronal loss from the entorhinal cortex, amygdala, and hippocampus2.
The pathology of AD is also characterized by two pathognomonic entities, intracellular neurofibrillary tangles (NFTs) and extracellular plaques.
The major constituent of plaques is a collection of peptides called beta-amyloid (Aβ) while NFTs are largely composed of paired helical filaments of an abnormally hyperphosphorylated form of the microtubule associated protein tau (MAPT or tau).
Tau pathology spreads in a predictable manner, from the allocortex to the association areas of the neocortex and lastly the primary cortices3,4.
Cortical layers within the entorhinal cortex from severely affected subjects can exhibit up to 90% neuronal loss5 while regions such as the primary visual cortex remain essentially unaffected by the disease process with mild Aβ accumulation and minor alterations in neuronal form and functions.
The popular amyloid cascade hypothesis (ACH), suggests that the common sporadic forms of AD results from the accumulation of soluble oligomeric forms of Aβ in the neuropil.
These toxic products disrupt neuronal kinase/phosphatase and redox balance, leading to tau hyperphosphorylation, NFT formation and neurodegeneration6.
However, the mechanisms leading to the accumulation of Aβ oligomers in sporadic AD are not clear.
Epidemiological studies have not been particularly informative with only ageing, the possession of the apolipoprotein E (APOE) ε4 allele, female gender and diabetes being consistently identified as risk factors7.
In addition to plaques and NFTs, the AD brain is also characterized by oxidative damage to proteins, nucleic acids8 and lipids9.
It is generally accepted that Aβ may act directly as a pro-oxidant10 or indirectly by precipitating NMDA receptor-dependent Ca2+ influxes that result in mitochondrial dysfunction and the subsequent generation of reactive oxygen species (ROS)11.
These, in turn, stimulate antioxidant response pathways that involve redox sensitive transcription factors e.g., NRF-2 (nuclear factor, erythroid 2-like 2; encoded by NFE2L2).
Activation of NRF-2 is known to stimulate the potent inducible antioxidant hemeoxygenase-1 (HO-1) and possibly other endogenous antioxidant response elements11.
Yet other researchers hypothesise that Aβ accumulation may be a consequence of oxidative stress12 and suggest that Aβ13 and indeed tau14 act as anti-oxidants in AD (reviewed by Sutherland et al.)15 thereby, potentially limiting the observed radical-mediated damage to DNA, proteins and lipids that occurs during the pathogenesis of this neurodegenerative disease16,17.
Notwithstanding that the outcomes from clinical trials of Aβ-modifying therapies in asymptomatic individuals are outstanding, there is still a need to pursue adjunctive or alternative therapeutic targets in AD, through accurate modelling of the early phases of the disease18.
In this study, we explore markers of oxidative damage in the superior temporal gyrus (STG). The AD-STG at post-mortem shows moderate levels of insoluble tau and plaques while retaining the majority of its neurons19.
Therefore, the post-mortem STG may represent a surrogate for the state of the most severely affected regions such as the entorhinal cortex, earlier in the disease course.
An understanding of events in the post-mortem STG may allow a better temporal appreciation of linkages between oxidative stress and AD pathogenesis and more generally reveal novel pathways to exploit in the development of therapeutics to combat AD at a stage prior to irreversible cognitive decline.
Source:
Johns Hopkins University
Media Contacts:
Rachel Butch – Johns Hopkins University
Image Source:
The image is credited to Chirag Vasavda.
Original Research: Closed access
“Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide”. Chirag Vasavda et al.
Cell Chemical Biology. doi:10.1016/j.chembiol.2019.07.006



{kind=link}
[…] Bilirubin appears to have neuroprotective properties from oxidative stress damage […]