











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A Melbourne led study has uncovered a new neurodegenerative disorder in which children experience developmental regression and severe epilepsy.
The study, led by the Murdoch Children’s Research Institute (MCRI) and published in the American Journal of Human Genetics, found a variation in a gene causes a severe childhood-onset neurodegenerative disorder that has never before been described.
MCRI and Victorian Clinical Genetics Services (VCGS) Associate Professor Sue White said this newly discovered condition was different from other chronic neuroinflammation implicated in other neurodegenerative conditions such as Alzheimer’s disease, Parkinson’s disease and frontotemporal dementia.
Associate Professor White said the study participants started with normal or mild developmental delay, and the onset of seizures started within the first year of life.
All had a severe and progressive developmental regression following a seizure, she said.
The study looked at six children from four families with the gene variant who had a similar degenerative condition, the cause of which was unlocked by genomic testing.
The genomic testing and data analyses were conducted for four participants at VCGS in Melbourne and one each at Ospedale Pediatrico Bambino Gesù in Italy and at Baylor College of Medicine in the US.
Associate Professor White said the disorder, with features suggestive of neuroinflammation, appeared to require two copies of the defective gene, meaning both parents had to be carriers of one altered copy.
“In our study the same gene variant was identified in three children of the same ethnic background,” she said.
“While the families do not report that their two families are directly related, they are presumed to be distantly related due to the overlap of their family histories, with common ancestors originating from the same town.”
The researchers used advanced molecular techniques to dissect the likely cellular pathway affected by the mutation in the NRROS gene. By inserting the gene into cells in the laboratory, they identified other molecules that NRROS interacts with.
These molecules are crucial for a number of brain cell functions, including adding the insulating layers around nerve fibres, and producing brain immune cells.
“In line with these laboratory findings, our study participants had neurodegenerative symptoms with difficult to control epilepsy, developmental regression, and delayed myelination,” Associate Professor White said.
“The myelination process is vitally important to healthy central nervous system functioning, enabling nerve cells to transmit information faster and allows for more complex brain processes.”
MCRI Professor John Christodoulou said the outcomes of this research highlighted the power of new genomic sequencing technologies that had ended a diagnostic odyssey that for some families may take years.
“Now that we know the causative gene, we are in a better position to understand the underlying biology behind the disorder, which we hope in future may translate to targeted treatments specific for the disorder,” he said.
Microglia are tissue-resident macrophages playing essential roles in central nervous system (CNS) development and homeostasis [14, 17]. The importance of microglia for brain health in humans has been highlighted by the definition of Mendelian disorders associated with dysfunction of microglia-related proteins.
These so-called microgliopathies [20] comprise a diverse set of neurological phenotypes including disease due to mutations in CSF1R [5, 8, 13], DAP12 and TYROBP/TREM2 [4, 7], USP18 [3, 11, 16, 18], and IRF8 [2, 6]. Here, we describe a novel early onset lethal encephalopathy due to mutations in the microglial-associated protein NRROS.
We ascertained three patients demonstrating a stereotyped clinical and neuroradiological phenotype (Supplementary material). Patients 1 (P1) and P2, both females, were the first and third children of non-consanguineous parents of Maori descent (family F1442), whilst P3 (family F2382), a male, was the first child of first cousin south Asian parents (Supplementary Figure 1).
All three children were born after a normal pregnancy and delivery, and early development was unremarkable. However, in the second year of life, they experienced the onset of refractory seizures and neurodegeneration, leading to death between the ages of 27 and 36 months. Metabolic testing, including for mitochondrial dysfunction, was non-contributory.
Neuroimaging initially demonstrated fine calcification at the depths of the cerebral gyri, with normal white matter (Fig. 1). As disease progressed, repeat imaging revealed increased calcification, severe generalized atrophy with ventricular dilatation, and diffuse signal changes in cerebral and cerebellar white matter.
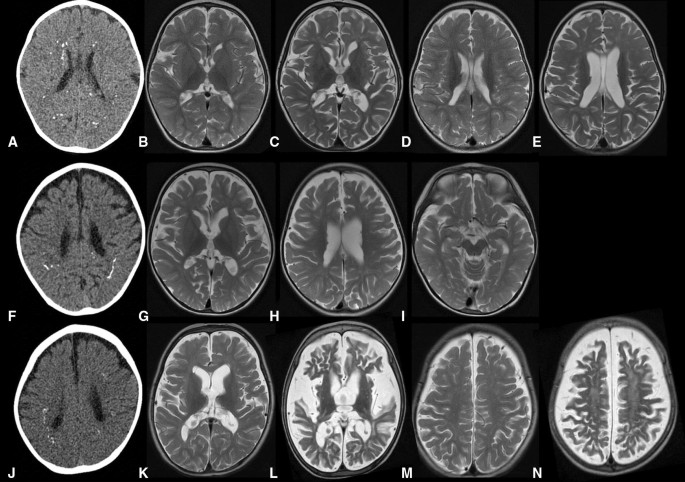
Exome sequencing identified homozygous NRROS variants in the affected children from both families: a c.1777C > T/p.(Gln593*) and a c.1257del/p.(Gly420AlafsTer14) in F1442 and F2382, respectively. Cellular material was not available from any of the patients.
However, both of these variants are predicted to result in a truncated protein, and both are very rare, with the p.(Gln593*) not previously recorded, and the p.(Gly420AlafsTer14) reported on only 1 of 251,438 alleles on gnomAD.
Detailed pathological examination was undertaken on P3, demonstrating abnormalities confined to the CNS. Gross examination indicated a significant cerebral atrophy (Supplementary Figure 2). Histologically, there was both grey and white matter pathology throughout the cerebrum, cerebellum, and brainstem. Focal calcification was noted in the neuropil.
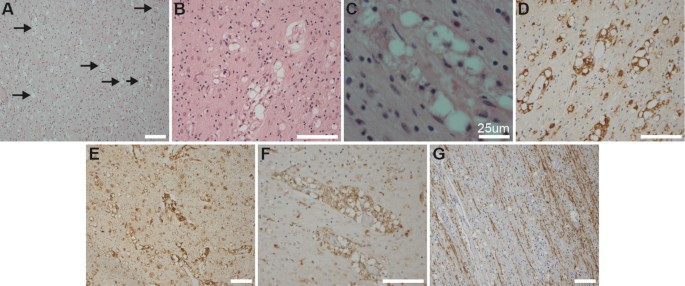
There was widespread neuronal loss with reactive gliosis throughout the grey matter (Supplementary Figure 3). The most striking pathological finding was the accumulation of foamy macrophages, predominantly in a perivascular distribution, throughout the white matter, extending from frontal to occipital white matter
(Fig. 2a–c), and through cerebellar white matter and descending corticospinal pathways in the basis pontis. These foamy cells immunoreacted with CD68, MHC Class II (CR3/43), and p22phox (Fig. 2c–f), but did not immunoreact with CD163, Iba1, NRROS, CD3, P2Y12, or TMEM119 (Supplementary Figure 3).
There was reduced myelin basic protein (MBP) expression in the white matter (Fig. 2g) compared to age-matched controls, although there was preservation of U fibers. Occasional axonal spheroids were noted, albeit this was not a prominent feature (Supplementary Figure 3).
We assessed the cellular expression of NRROS, and the mouse homolog Nrros (Lrrc33), in human and mouse brain respectively, by mining curated transcriptomic data sets (Supplementary Figure 4).
In fresh post-mortem human cortical microglia and brain samples, NRROS was highly expressed in isolated microglia, although less abundantly than established microglial signature genes.
The expression of NRROS was enriched > 50-fold in microglia compared to whole brain, indicating that microglia are the major cell type expressing NRROS in human brain parenchyma.
A similar pattern of highly enriched expression of Lrrc33/Nrros was observed in CD11b+ microglia/macrophages in mouse brain relative to brain extracts, and in microglia versus other parenchymal cell types. Comparison of parenchymal microglia with CNS perivascular macrophages (PVMs) showed significantly greater expression in the latter.
The clinical features observed in our patients recapitulate those in mice with Nrros/Lrrc33 deficiency. Nrros−/−mice exhibited progressive neurological decline, including motor defects and abnormal locomotor activity, from age 2–3 months and death by 6 months of age [15, 21].
Neuropathology in these mouse models includes neuronal loss, demyelination, axonal pathology, astrogliosis, and the increased presence of foamy macrophages, all of which were seen in our case. Of note, there was no indication of immune-mediated inflammation in our case or either of these mouse models.
NRROS is a leucine-rich repeat containing transmembrane protein localized to the endoplasmic reticulum, and preferentially expressed in myeloid cells. Reported functions include the regulation of reactive oxygen species (ROS) production through control of NOX2 stability [12], responsiveness of Toll-like receptor signaling [19], and processing/activation of transforming growth factor (TGF)-β via physical interactions with the latent complex [10, 15].
NRROS expression is restricted to microglia within the CNS parenchymal compartment in humans and mice. The present case showed disruption to the distribution, density, and cell morphology of IBA1 cells alongside loss of P2Y12 staining and weak TMEM119 immunoreactivity, indicative of marked parenchymal microglial abnormalities.
Both Nrros−/− mouse studies observed a loss of homeostatic gene expression profile which included suppression of P2ry12 and Tmem119 expression, and a shift towards a phenotype resembling PVMs [15, 21].
Although Nrros is expressed in peripheral mononuclear cells, a series of crosses and bone marrow transplant experiments showed a negligible contribution of peripheral macrophages to the onset of the Nrros−/− phenotype [21]. Of note, selective deletion of Nrros in microglia during pregnancy indicated a cell-intrinsic role for NRROS.
In contrast, Nrros deletion induced in 3-week-old mice did not cause neuropathological changes or neurological abnormalities [21], implying that NRROS is important during microglial establishment at embryonic/postnatal stages, but may be dispensable for maintenance of adult microglia.
Functions of NRROS proposed above, notably in ROS and TGFβ regulation, may be important in disease pathogenesis. p22phox was markedly up-regulated in PVMs in our case, suggesting that an absence of functional NRROS may result in increased p22phox-NOX2 binding, with the potential for increased superoxide radical formation.
However, a cross of Nrros−/− and Cybb−/− (encoding NOX2) mice did not rescue the Nrros−/− phenotype [21]. Mice with CNS or microglial-restricted disruption during development of other key nodes in the TGFβ activation/signaling pathway, including deletion of αVβ8 integrin or TGFBR2 [1], develop highly similar pathological, microglial, and neurological abnormalities to Nrros−/− mice. Moreover, human TGFβ1 loss-of-function mutations causing early onset leukoencephalopathy were described recently [9].
Taken together with the mouse data, our findings indicate that NRROS is indispensable in controlling the early development of a homeostatic microglial population and/or its ongoing preservation in the postnatal brain, thereby suggesting a loss of NRROS function as a novel microgliopathy in humans.
References
1. Arnold TD, Lizama CO, Cautivo KM, Santander N, Lin L, Qiu H et al (2019) Impaired αVβ8 and TGFβ signaling lead to microglial dysmaturation and neuromotor dysfunction. J Exp Med 216:900–915. https://doi.org/10.1084/jem.20181290
2. Bigley V, Maisuria S, Cytlak U, Jardine L, Care MA, Green K et al (2018) Biallelic interferon regulatory factor 8 mutation: a complex immunodeficiency syndrome with dendritic cell deficiency, monocytopenia, and immune dysregulation. J Allergy Clin Immunol 141:2234–2248. https://doi.org/10.1016/j.jaci.2017.08.044
3. Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L et al (2015) USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J 34:1612–1629. https://doi.org/10.15252/embj.201490791
4. Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N et al (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol 70:78–84. https://doi.org/10.1001/jamaneurol.2013.579
5. Guo L, Bertola DR, Takanohashi A, Saito A, Segawa Y, Yokota T et al (2019) Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-pyle disease spectrum and degenerative encephalopathy with brain malformation. Am J Hum Genet 104:925–935. https://doi.org/10.1016/j.ajhg.2019.03.004
6. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J et al (2011) IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 365:127–138. https://doi.org/10.1056/NEJMoa1100066
7. Konishi H, Kiyama H (2018) Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci 12:206. https://doi.org/10.3389/fncel.2018.00206
8. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK (2018) CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology 91:1092–1104. https://doi.org/10.1212/WNL.0000000000006642
9. Kotlarz D, Marquardt B, Barøy T, Lee WS, Konnikova L, Hollizeck S et al (2018) Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat Genet 50:344–348. https://doi.org/10.1038/s41588-018-0063-6
10. Ma W, Qin Y, Chapuy B, Lu C (2019) LRRC33 is a novel binding and potential regulating protein of TGF-β1 function in human acute myeloid leukemia cells. PLoS One 14:e0213482. https://doi.org/10.1371/journal.pone.0213482
11. Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD et al (2016) Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 213:1163–1174. https://doi.org/10.1084/jem.20151529
12. Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA et al (2014) NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 509:235–239. https://doi.org/10.1038/nature13152
13. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R et al (2019) Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet 104:936–947. https://doi.org/10.1016/j.ajhg.2019.03.010
14. Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312. https://doi.org/10.1038/nrn3722
15. Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J et al (2018) A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 174:156–171.e16. https://doi.org/10.1016/j.cell.2018.05.027
16. Rodero MP, Crow YJ (2016) Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med 213:2527–2538. https://doi.org/10.1084/jem.20161596
17. Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23:1018–1027. https://doi.org/10.1038/nm.4397
18. Schwabenland M, Mossad O, Peres AG, Kessler F, Maron FJM, Harsan LA et al (2019) Loss of USP18 in microglia induces white matter pathology. Acta Neuropathol Commun 7:106. https://doi.org/10.1186/s40478-019-0757-8
19. Su X, Mei S, Liang X, Wang S, Liu J, Zhang Y et al (2014) Epigenetically modulated LRRC33 acts as a negative physiological regulator for multiple Toll-like receptors. J Leukoc Biol 96:17–26. https://doi.org/10.1189/jlb.0813457
20. van der Knaap MS, Bugiani M (2017) Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta neuropathol 134:351–382. https://doi.org/10.1007/s00401-017-1739-1
21. Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA et al (2017) Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol 18:633–641. https://doi.org/10.1038/ni.3743
More information: Xiaomin Dong et al. Bi-allelic LoF NRROS Variants Impairing Active TGF-β1 Delivery Cause a Severe Infantile-Onset Neurodegenerative Condition with Intracranial Calcification, The American Journal of Human Genetics (2020). DOI: 10.1016/j.ajhg.2020.02.014



{kind=link}