











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



The first sign of trouble for a patient with a growing brain tumor is often a seizure.
Such seizures have long been considered a side effect of the tumor. But now a joint team of Columbia engineers and cancer researchers studying brain tumors has found evidence that the seizures caused by an enlarging tumor could spur its deadly progression.
These interactions, described today in Cell Reports, were revealed using a novel mouse brain imaging technology that tracks real-time changes in brain activity and blood flow as a tumor grows in the brain.
The research identifies potential new targets for diagnosing and treating glioma, a rare but aggressive form of brain cancer, notable in recent years for having claimed the lives of United States Senator John McCain and Beau Biden, the son of Vice President Joe Biden.
“As gliomas spread within the brain, they gradually infiltrate surrounding brain regions, altering blood vessels and interactions between neurons and other brain cells,” said Peter Canoll, MD, PhD, professor of pathology and cell biology at Columbia’s Vagelos College of Physicians and Surgeons and the paper’s co-senior author.
“Neuro-oncologists have generally focused on developing ways to selectively kill glioma cells, but we are also interested in understanding how infiltrating glioma cells change the way that the brain functions. We believe that this approach can lead to new treatments for this terrible disease.”
Meanwhile, Elizabeth Hillman, PhD, a professor of biomedical engineering at Columbia’s School of Engineering and Applied Science, had developed a novel method for real-time imaging of both neural activity and blood flow dynamics in the brains of mice.
Called wide-field optical mapping, or WFOM, Dr. Hillman and her lab were using the system to study how neuronal activity in the brain drives local changes in blood flow, a process known as neurovascular coupling.
Drs. Hillman and Canoll realized that combining Dr. Hillman’s imaging platform with Dr. Canoll’s method of generating realistic tumors in the brain of mice, could let them explore how brain activity was affected during tumor growth.
“We used WFOM to image the brains of mice every few days for many weeks, observing how tumors grew and invaded different areas,” said Dr. Hillman, who is also a Zuckerman Institute Principal Investigator.
“We studied mice whose neurons were labeled with a green fluorescent calcium indicator, which gets brighter when neurons are more active, letting us detect how tumor invasion affected the normal activity of neurons and dilations and constriction of blood vessels.”
The team first found that migrating glioma cells desynchronized both neuronal activity and blood flow changes that normally fluctuate together across either side of the brain.
They also found that the tumor affected neurovascular coupling — making blood vessels less likely to dilate and provide fresh blood when neurons fired.
But another thing also caught the researchers’ eye.
“We saw some flashes in our images of neuronal activity, accompanied by big changes in blood flow,” said Dr. Hillman. “When we looked more closely, we found these flashes became more and more frequent as the tumors grew, and in some cases we saw massive, profound blasts of neuronal activity.”
“We realized that as the tumors progressed in the brains of mice, we were seeing many seizure-like discharges, which eventually progressed into full-blown seizures that resemble the generalized seizures that glioma patients often experience,” said Dr. Canoll.
“We noticed that these seizures were most prominent at the edges of the tumor, where tumor cells were growing into and intermingling with surrounding healthy brain.”
During these generalized seizures, WFOM also revealed that blood oxygenation levels within the tumor dropped sharply.
This finding was surprising, and concerning, as tumor cells are known to thrive in low-oxygen, or hypoxic, environments.
“When brain tissue becomes hypoxic, brain cells can secrete proteins that could actually stimulate tumor growth, migration, proliferation and progression,” said Dr. Canoll.
“We think that the altered neurovascular coupling in the tumor is causing hypoxia during seizures, and might create a vicious cycle of tumor growth, seizure, hypoxia and further tumor growth.”
The team’s findings point to a number of promising targets to disrupt glioma’s vicious assault on the brain.
“To break this cycle of tumor growth, we could target reducing seizures. We may be able to use WFOM to determine which types of drug are most effective for suppressing these types of seizure, while not interfering with cancer treatments,” said Dr. Canoll.
“We will also be looking to see whether small seizure events that might be difficult for a patient to perceive could be an early warning sign of tumor development or regrowth.”
What the team learned could also help with diagnosis and surgical guidance.
“Functional magnetic resonance imaging, fMRI, is a human brain imaging method that detects active regions via local changes in brain blood flow,” said Dr. Hillman, who is also a professor of radiology at Columbia’s Vagelos College of Physicians and Surgeons.
“Our results suggest that fMRI may be unreliable for guiding surgeons to avoid specific brain regions, if the tumor alters blood flow changes.
However, the changes in synchrony and neurovascular coupling that we observed could also potentially be leveraged as biomarkers to detect tumor regions.”
The team’s results also highlight the power of interdisciplinary collaborations. Dr. Hillman developed the study’s imaging and analysis techniques with support from the NIH BRAIN Initiative, and the team combined them with novel mouse glioma models developed in the Canoll lab to study alterations in brain function associated with neurological disease.
“We are so excited to demonstrate that these methods can give us a new view of how diseases affect how the brain works,” said Dr. Hillman.
“We hope this study prompts more scientists and clinical researchers to start using in-vivo imaging methods to advance our understanding of brain diseases and disorders.”
This paper is titled “Glioma-induced alteration in neuronal activity and neurovascular coupling during disease progression.”
Additional contributors include the three co-first authors Mary Katherine Montgomery, Sharon Kim, PhD, and Athnassios Dovas, PhD, and the other co-authors Hanzhi Zhao, Alexander Goldberg, Weihao Xu, Alexis Yagielski, Morgan Cambareri, Kripa Patel, Angeliki Mela, PhD, Nelson Humala, David Thibodeaux, Mohammed Shaik, Ying Ma, Jack Grinband, PhD, Daniel Chow and Catherine Schevon, MD, PhD.
Funding: This research was supported by the BRAIN Initiative (RF1 MH115276), the National Institutes of Health (R01NS076628, R01NS063226, U01CA236554, R03 NS090151-01), the National Center for Advancing Translational Sciences (CTSA-pilot funding UL1 TR000040), Columbia ROADS (RG31), the James F. McDonnell Foundation and the American Epilepsy Society.
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive primary malignant brain tumour and accounts for 60% of brain tumours in adults (1). The global incidence of GBM is <10 per 100,000 persons (2) and has increased over the last decade (3). GBM patients have a poor prognosis with a 1-year survival rate of 37.2%, a 5-year survival rate of 5.1% (4) and a median survival of ~10 months (5).
GBM is divided into three subgroups based on isocitrate dehydrogenase 1 (IDH1) and IDH2 mutation status: IDH-mutant, IDH-wild-type and NOS (not otherwise specified) (6–8). However, despite this classification, the majority of GBM patients receive identical treatments, and few targeted therapies currently exist, contributing to the poor outcomes typically experienced by GBM patients.
This review will outline the current treatment options for GBM and discuss some of the more recent developments in targeted therapies being investigated for the treatment of GBM.
Current Treatment Options
The treatment of brain tumours faces unique challenges, most notably the presence of the blood brain barrier (BBB), a highly selective semipermeable barrier that separates blood from the brain.
The BBB is comprised of the endothelial cells of capillaries, astrocytes surrounding the capillary, and pericytes embedded in the capillary basal lamina. Physiochemical properties including molecular weight, lipophilicity and charge affect the ability of a molecule to cross the BBB (9).
The BBB prevents nearly all large molecules (>400 Da) (10) and ~98% of small molecule drugs from entering the central nervous system (CNS) (11). The current treatment pipeline begins with surgical resection of the tumour, if applicable and safe to do so, followed by radiotherapy and concomitant chemotherapy (12, 13).
The initial therapeutic approach for GBM is surgery, where maximal resection is associated with longer progression-free survival (PFS) and overall survival (OS) (14). Resection is not a curative approach; hence, patients typically undergo radiotherapy and chemotherapy as an adjunct (15).
Radiotherapy, at a total dose of 60 Gy, is administered as either primary treatment or following surgery (16), both resulting in improvements to PFS and OS. Concomitant administration of temozolomide [150–200 mg/m2/day for 5 days each 28-days cycle (12, 13)], an oral alkylating agent, significantly increases OS in patients with newly diagnosed GBM from 12.1 months with radiotherapy alone to 14.6 months with radiotherapy and temozolomide (13).
However, despite this increase in survival with radiotherapy and temozolomide, tumour progression and recurrence typically occur (17, 18), due to the development of resistance to temozolomide (19, 20). Once GBM recurrence occurs, therapeutic options for patients are limited (21, 22).
Recently, tumour treating fields (TTFields; Optune), which deliver electric fields to the tumour location to disrupt cancer cell division, have emerged as an FDA-approved treatment for both recurrent and newly diagnosed GBM (23). However, the identification of new targets to facilitate the development of novel targeted therapies is warranted.
Emerging Targeted Therapies
GBM is an invasive tumour with hallmarks of neoangiogenesis and intratumour heterogeneity, contributing to the poor prognosis observed (24). A variety of genetic and epigenetic alterations have been identified in GBM that influence patient prognosis (Table 1).
Despite this heterogeneity, a large-scale analysis of genetic aberrations in GBM identified three main signalling pathways that are commonly dysregulated: activation of the receptor tyrosine kinase (RTK)/Ras/phosphoinositide 3-kinase (PI3K) pathway (88%), inhibition of p53 (87%), and retinoblastoma protein (Rb) signalling pathways (78%) (34). Drugs targeting many of these commonly observed alterations have been investigated as potential targeted therapies for GBM.
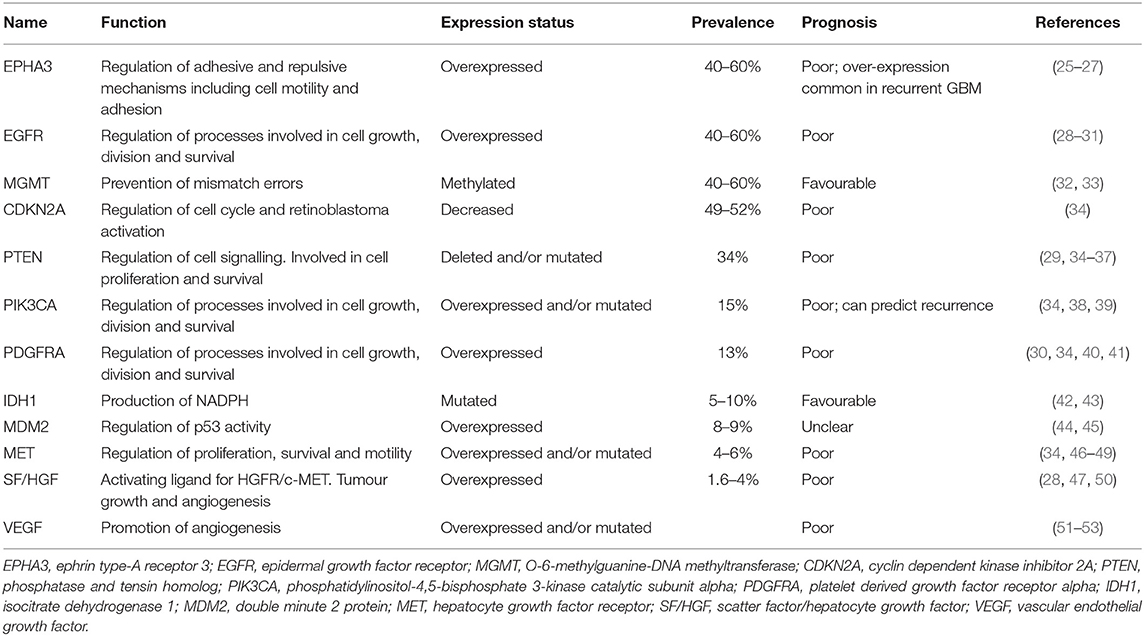
EphA3 Receptor Inhibitors
The EphA3 receptor is overexpressed in 40–60% of GBM tumours and is commonly overexpressed in recurrent GBM (Table 1) (25). EphA3 is highly expressed on the tumour-initiating cell population in glioma, maintains tumour cells in a less differentiated and stem cell-like state, and its expression mediates the tumourigenic potential in GBM cells in vitro (26), suggesting that EphA3 may be a potential target for the treatment of GBM (Table 2).
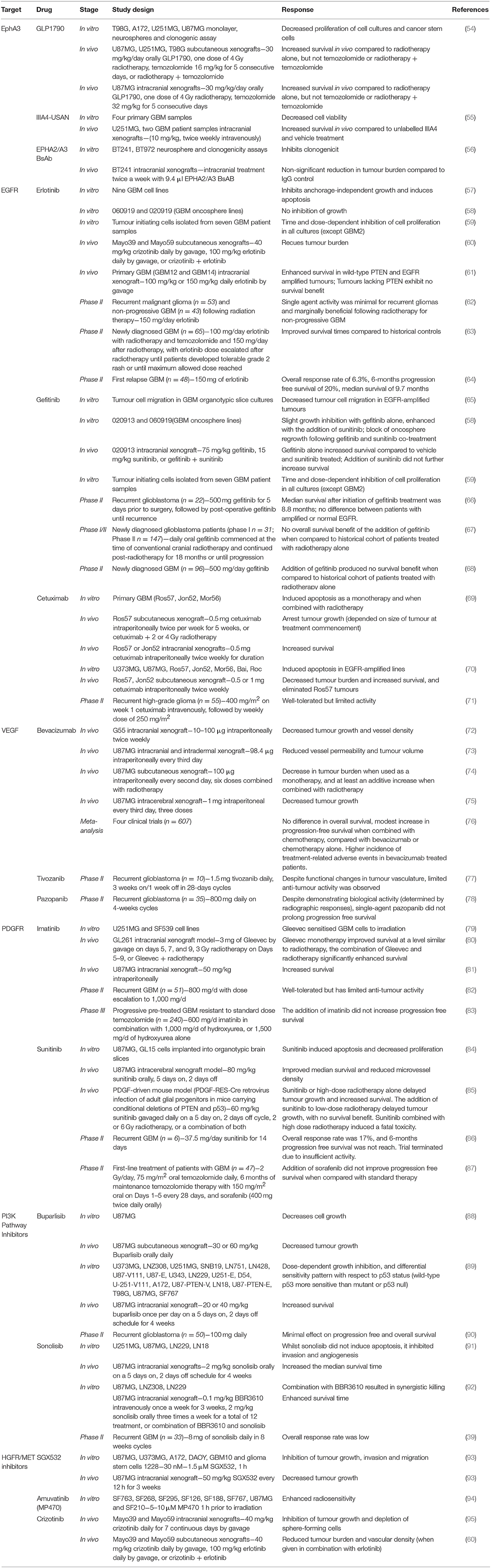
A small molecule inhibitor of the EphA3 receptor, GLPG1790, has demonstrated superior tumour reduction in U251MG and U87MG subcutaneous xenograft models when compared to radiotherapy alone, however, GLPG1790 was not as effective as treatment with radiotherapy and concomitant temozolomide (54).
Whilst GLPG1790 did not exhibit improved benefit over the current therapies, additional strategies for targeting EphA3 are being examined. An EphA3 monoclonal antibody (IIIA4) that binds the EphA3 globular ephrin-binding domain has been developed, and the humanised version (ifabotuzumab) is the subject of an investigator-sponsored Phase 0/1 clinical trial currently underway in patients with recurrent GBM (55) to identify the optimal dose for tumour penetration.
The IIIA4 antibody conjugated to the cytotoxic microtubule-targeting agent maytansine (IIIA4-USAN), induced apoptosis in four primary GBM cell lines in vitro, and significantly increased survival in an orthotopic model in vivo (55). Providing further evidence for the potential suitability of monoclonal antibodies targeting EphA3, a bispecific antibody against EphA2/A3 reduced clonogenicity in vitro and decreased tumour burden in vivo (56).
Taken together, these studies indicate that EphA3 receptor inhibitors may be promising treatments for EphA3 receptor-amplified GBM, including recurrent disease, however, this remains to be tested in the clinic.
EGFR Inhibitors
Epidermal Growth Factor Receptor (EGFR) amplifications and mutations are detected in 40–60% of GBM cases (28, 96) (Table 1) and are generally indicative of poor prognosis (97). EGFR (also referred to as ERBB1 or HER1) is a member of the HER superfamily of RTKs, along with ERBB2, ERBB3, and ERBB4. Binding of a ligand to the ligand-binding site of these receptors induces receptor homo- or heterodimerisation, producing a conformational change that activates the intracellular tyrosine kinase domain.
This results in autophosphorylation of the cytoplasmic tail and induces a variety of downstream signalling pathways. The overexpression or mutation of EGFR leads to downstream signalling that impairs apoptosis, enhances proliferation, and angiogenesis. The most common mutant form found in GBM is ΔEGFR (EGFRvIII, or de2-7EGFR), arising through an 801 base pair in-frame deletion from the extracellular domain (98). Due to the high incidence of EGFR amplifications, a variety of EGFR inhibitors have been examined both pre-clinically and clinically (Table 2).
Small Molecule Inhibitors
Small molecule tyrosine kinase inhibitors are the most widely studied EGFR inhibitors in GBM, and include erlotinib, gefitinib, and lapatinib. Erlotinib inhibits anchorage-independent growth of GBM cells in vitro in an EGFR expression-dependent manner and induces greater levels of apoptosis in more malignant GBM phenotypes (57).
The tumour-initiating cell population, which is resistant to radiotherapy (99), is sensitive to erlotinib in a phosphatase and tensin homolog (PTEN) and Akt dependent manner (59), suggesting that erlotinib may eliminate this population in vivo. Further, treatment with erlotinib was shown to reduce tumour burden in two GBM patient-derived xenograft (PDX) models (60).
However, further studies using additional GBM PDX models demonstrated that tumours overexpressing EGFR were only sensitive to erlotinib if they also expressed PTEN (61). As PTEN expression is downregulated in ~34% of GBM patients (29), this indicates that erlotinib may not be a suitable treatment for the majority of GBM patients overexpressing EGFR. Indeed, erlotinib was not effective as a monotherapy in recurrent GBM patients and was only marginally beneficial following radiotherapy for non-progressive GBM patients (62).
Despite a limited number of complete and partial responses in a Phase II study in first-relapse GBM, the 6-months PFS and median survival was similar to that previously reported for patients undergoing chemotherapy (64), suggesting that erlotinib may be useful in this setting. However, due to the non-randomised nature of this trial, these results must be interpreted cautiously. By contrast, improved survival (19.3 vs. 14.1 months) was observed when combined with temozolomide and radiotherapy (63), suggesting that erlotinib may be beneficial when combined with other treatments, rather than as a monotherapy.
In contrast to erlotinib, gefitinib exhibits anti-tumour activity independent of the expression level of EGFR (100). Gefitinib inhibits GBM cell migration (65), reduces proliferation of human glioma tumour-initiating cells in vitro (59) and enhances survival in an intracranial GBM mouse xenograft model in vivo (58).
Taken together, these pre-clinical studies indicate that gefitinib may be clinically beneficial. However, despite gefitinib reaching high concentrations in GBM tumour tissue (22-fold higher compared to plasma) and the significant dephosphorylation of EGFR achieved (66), limited clinical effects have been observed in Phase II trials. Several Phase I/II studies have demonstrated that whilst the addition of gefitinib to radiotherapy is well-tolerated, it has no survival benefit (67, 68).
Monoclonal Antibodies
Although tumour immunotherapy has shown some success for the treatment of melanoma and haematological cancers, the applicability to GBM presents more of a challenge. Monoclonal antibodies directed against wild-type EGFR and ΔEGFR have been developed, with the best characterised in GBM being cetuximab. Pre-clinical studies have shown that treatment with cetuximab alone and in combination with radiotherapy increases survival in vivo (69) and can also completely eliminate tumours in EGFR-amplified PDX models (70). A phase II trial examining cetuximab treatment in patients with recurrent high-grade glioma showed that cetuximab was well-tolerated, but exhibited limited activity in this patient population (71).
VEGF Inhibitors
With the largely disappointing clinical results for EGFR inhibitors, additional targets are being investigated, including vascular endothelial growth factor (VEGF), which is highly expressed in glioma cells. High VEGF expression is directly associated with the poor prognosis and malignancy of gliomas (51–53, 101). VEGF is a dimeric polypeptide that binds to the VEGF receptors 1 (VEGFR-1) and VEGFR-2, and the co-receptors neuropilin 1 and 2. Following this interaction, VEGF mediates angiogenesis and cell proliferation.
Under hypoxic conditions, hypoxia-inducible transcription factors translocate to the nucleus which activate VEGF leading to increased angiogenesis in an attempt to counteract hypoxia (102). GBM tumours are commonly hypoxic and have increased VEGF expression that contributes to the irregular vasculature in GBM, prompting the investigation of VEGF as a potential therapeutic target (Table 2).
Small Molecule Inhibitors
Several VEGF inhibitors have been examined for the treatment of GBM, including the small molecule inhibitors, tivozanib, and pazopanib. Phase II studies of tivozanib (77) and pazopanib (78) in recurrent glioblastoma showed that these inhibitors exhibited limited anti-tumour activity and did not prolong PFS in this patient population. These trials highlight the limitations of anti-VEGF monotherapy.
Monoclonal Antibodies
Bevacizumab, a humanised monoclonal antibody against VEGF, blocks angiogenesis and thereby reduces tumour growth in a variety of GBM mouse models as a monotherapy and when combined with radiotherapy (72–75). These promising pre-clinical studies led to the clinical investigation and subsequent approval of bevacizumab for the treatment of recurrent GBM (103).
However, a meta-analysis of four clinical trials including 607 patients demonstrated that the addition of bevacizumab to standard chemo-radiotherapy in the upfront setting only improves PFS, with no improvement in OS, but with an increase in the number of treatment-related adverse events (76). Of note, a decline in neurocognitive function is more frequently observed following bevacizumab treatment, as bevacizumab impairs hippocampal synaptic plasticity and decreases dendritic spine number and length (104). The modest treatment responses combined with the increased treatment-related adverse events raises concerns about the suitability of the use of bevacizumab as a treatment for GBM.
Inhibition of Multiple RTKs
Multiple RTKs are coactivated in GBM tumours (105), introducing redundancy and limiting the efficacy of therapies targeting single RTKs. EGFR and platelet derived growth factor receptor A (PDGFRA) protein co-expression occurs in 37% of GBM (106). PDGFRA is the second most frequently amplified (10–13%) receptor tyrosine kinase in GBM (28, 107) (Table 1). A variety of multi-RTKs inhibitors have been examined both pre-clinically and clinically (Table 2).
Imatinib is a small molecule that inhibits PDGFRA and PDGFRB, as well as the RTKs c-Abl and c-Kit, and is a radiation-sensitising agent for glioma cells in vitro (79) and in orthotopic GBM models in vivo (80, 81). These promising pre-clinical studies led to the initiation of clinical trials. However, whilst imatinib was well-tolerated in recurrent GBM patients, it exhibited limited anti-tumour activity (82).
Subsequent Phase III trials have examined imatinib in combination with hydroxyurea, rather than as a monotherapy (83). This trial showed that there was no PFS benefit to the addition of imatinib to hydroxyurea, or hydroxyurea alone. Further, a recent study has demonstrated that imatinib treatment can increase GBM cell migration and invasion in vitro (108), providing further potential insight as to why imatinib has failed in clinical trials for GBM.
Following the failure of imatinib in clinical trials, additional multi-RTK inhibitors have been studied. Sunitinib is an oral, small molecule that inhibits PDFR and VEGFR, and thereby reduces vascularisation and triggers apoptosis to produce tumour reduction. In pre-clinical models, sunitinib treatment induced apoptosis in vitro, and improved survival in an intracerebral GBM mouse model (84).
Further, sunitinib treatment delayed tumour growth and increased survival in a PDGFF-driven mouse model, both as a monotherapy and in combination with low dose radiotherapy (85). By contrast, a Phase II trial and systematic review of the literature indicated that compared to conventional chemotherapy or bevacizumab, sunitinib has limited clinical activity in recurrent GBM as a monotherapy (86) or when combined with temozolomide or radiotherapy and temozolomide as a first-line treatment of patients with GBM (87).
PI3K Pathway Inhibitors
Genetic aberrations in GBM, including EGFR, PDGFRA, PTEN, TP53, and PIK3CA, drive the dysfunction of signalling pathways, including PI3K/Akt/mTOR, p53, and Rb1 (34). The PI3K/Akt signalling pathway is activated in most GBMs and plays a critical role in the regulation of signal transduction, and mediates a variety of cellular processes, including proliferation, survival, migration and angiogenesis in GBM.
The PI3K/Akt pathway is typically initiated via the activation of RTKs or G protein-coupled receptors, were the conformational changes in the C-terminal kinase domain produced by autophosphorylation provides binding sites for the regulatory subunits of PI3K, which leads to elevated lipid kinase activity of PI3K, and activation of Akt.
Despite the limited clinical efficacy of the previously described RTK inhibitors, as activation of each of these receptors leads to downstream activation of the PI3K/Akt pathway, it has therefore been suggested that PI3K pathway inhibitors may be beneficial in GBM. More than 50 PI3K inhibitors have been designed and are under investigation as treatments for a range of cancers. Several PI3K inhibitors have demonstrated pre-clinical efficacy in GBM (Table 2) and have entered into clinical trials for GBM treatment.
Buparlisib, a pan PI3K inhibitor, reduces GBM cell growth both in vitro and in vivo (88, 89). Buparlisib is the most frequently used PI3K inhibitor in clinical trials for GBM treatment, as it is well-tolerated and BBB permeable. However, single agent efficacy in Phase II trials in recurrent glioblastoma has been minimal (90). The lack of clinical efficacy was explained by incomplete blockade of the PI3K pathway in the tumour tissue. Whilst buparlisib showed minimal single-agent efficacy, the study of other PI3K inhibitors that achieve more-complete pathway inhibition may still be warranted.
Sonolisib is an irreversible wortmannin analogue that demonstrates a more persistent inhibitor effect on PI3K than wortmannin. Sonolisib inhibits invasion and angiogenesis in GBM cell lines in vitro and extends survival benefit in orthotopic xenograft models in vivo (91, 92). Despite these promising pre-clinical results, the response rate to sonolisib in a Phase II study in patients with recurrent GBM was low, and the study did not meet its primary endpoint (109).
HGFR/MET Inhibitors
Brain tumours secrete scatter factor (SF)/hepatocyte growth factor (HGF), the activating ligand for HGFR/MET (50), which has been associated with poor prognosis for GBM patients (Table 1) (110, 111). HGF is overexpressed in 1.6–4% of GBM patients, and via activation of MET, enhances tumour growth and angiogenesis (28). The MET proto-oncogene encodes for MET, an RTK that is overexpressed in 4–6% of GBM patients. A mutated, constitutively active variant of MET, METΔ7−8, has been identified in 6% of patients with high grade gliomas, including GBM, enhancing downstream signalling to promote tumour progression and angiogenesis (46).
SGX-523 is a small molecule inhibitor of HGFR/MET tyrosine kinase activity that inhibits tumour cell growth, migration and invasion in a panel of glioma cells in vitro and reduced tumour growth in a murine xenograft model of GBM using the U87MG GBM cell line (Table 2) (93). Additionally, amuvatinib (MP470) is a small molecule inhibitor that acts on multiple tyrosine kinases, including MET, has been shown to radiosensitise GBM cell lines both in vitro and in vivo (Table 2) (94).
Another small molecule inhibitor of MET kinase activity, crizotinib, inhibits the growth, sphere-forming capacity and expression of stem cell markers in a subcutaneous xenograft model of GBM using the U87MG cell line (95). However, in a subcutaneous xenograft model using Mayo39 and Mayo59 GBM cell lines, crizotinib was only effective at reducing tumour burden and vascular density when used in combination with the EGFR inhibitor erlotinib (60).
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. (2010) 60:277–300. doi: 10.3322/caac.20073
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Iacob G, Dinca EB. Current data and strategy in glioblastoma multiforme. J Med Life. (2009) 2:386–393.
PubMed Abstract | Google Scholar
3. Dobes M, Khurana VG, Shadbolt B, Jain S, Smith SF, Smee R, et al. Increasing incidence of glioblastoma multiforme and meningioma, and decreasing incidence of Schwannoma (2000–2008): findings of a multicenter Australian study. Surg Neurol Int. (2011) 2:176. doi: 10.4103/2152-7806.90696
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. (2015) 17(Suppl. 4):iv1–62. doi: 10.1093/neuonc/nov189
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Zhu P, Du XL, Lu G, Zhu JJ. Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: a population-based study. Oncotarget. (2017) 8:44015–31. doi: 10.18632/oncotarget.17054
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Kleihues P, Ohgaki H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro Oncol. (1999) 1:44–51. doi: 10.1093/neuonc/1.1.44
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. (2013) 19:764–72. doi: 10.1158/1078-0432.CCR-12-3002
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Kakee A, Terasaki T, Sugiyama Y. Brain efflux index as a novel method of analyzing efflux transport at the blood-brain barrier. J Pharmacol Exp Ther. (1996) 277:1550–9.
PubMed Abstract | Google Scholar
10. Gan HK, van den Bent M, Lassman AB, Reardon DA, Scott AM. Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat Rev Clin Oncol. (2017) 14:695–707. doi: 10.1038/nrclinonc.2017.95
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Xu YY, Gao P, Sun Y, Duan YR. Development of targeted therapies in treatment of glioblastoma. Cancer Biol Med. (2015) 12:223–37. doi: 10.7497/j.issn.2095-3941.2015.0020
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. (2016) 20:S2–8. doi: 10.1188/16.CJON.S1.2-8
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Allahdini F, Amirjamshidi A, Reza-Zarei M, Abdollahi M. Evaluating the prognostic factors effective on the outcome of patients with glioblastoma multiformis: does maximal resection of the tumor lengthen the median survival? World Neurosurg. (2010) 73:128–34; discussion: e16. doi: 10.1016/j.wneu.2009.06.001
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Vogelbaum MA. Does extent of resection of a glioblastoma matter? Clin Neurosurg. (2012) 59:79–81. doi: 10.1227/NEU.0b013e31826b2e75
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Perkins A, Liu G. Primary brain tumors in adults: diagnosis and treatment. Am Fam Physician. (2016) 93:211–7.
PubMed Abstract | Google Scholar
17. Sathornsumetee S, Rich JN. New approaches to primary brain tumor treatment. Anticancer Drugs. (2006) 17:1003–16. doi: 10.1097/01.cad.0000231473.00030.1f
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gallego O. Nonsurgical treatment of recurrent glioblastoma. Curr Oncol. (2015) 22:e273–81. doi: 10.3747/co.22.2436
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Happold C, Roth P, Wick W, Schmidt N, Florea AM, Silginer M, et al. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J Neurochem. (2012) 122:444–55. doi: 10.1111/j.1471-4159.2012.07781.x
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Felsberg J, Rapp M, Loeser S, Fimmers R, Stummer W, Goeppert M, et al. Prognostic significance of molecular markers and extent of resection in primary glioblastoma patients. Clin Cancer Res. (2009) 15:6683–93. doi: 10.1158/1078-0432.CCR-08-2801
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. (2017) 127:415–26. doi: 10.1172/JCI89587
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. (2017) 18:e315–29. doi: 10.1016/S1470-2045(17)30194-8
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Branter J, Basu S, Smith S. Tumour treating fields in a combinational therapeutic approach. Oncotarget. (2018) 9:36631–44. doi: 10.18632/oncotarget.26344
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Yi Y, Hsieh IY, Huang X, Li J, Zhao W. Glioblastoma stem-like cells: characteristics, microenvironment, and therapy. Front Pharmacol. (2016) 7:477. doi: 10.3389/fphar.2016.00477
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Ferluga S, Tomé CM, Herpai DM, D’Agostino R, Debinski W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget. (2016) 7:59860–76. doi: 10.18632/oncotarget.10978
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. (2013) 23:238–48. doi: 10.1016/j.ccr.2013.01.007
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Wykosky J, Gibo DM, Debinski W. A novel, potent, and specific ephrinA1-based cytotoxin against EphA2 receptor expressing tumor cells. Mol Cancer Ther. (2007) 6:3208–18. doi: 10.1158/1535-7163.MCT-07-0200
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
PubMed Abstract | CrossRef Full Text | Google Scholar
29. An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. (2018) 37:1561–75. doi: 10.1038/s41388-017-0045-7
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. (2015) 15:302–10. doi: 10.1038/nrc3918
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Hu X, Miao W, Zou Y, Zhang W, Zhang Y, Liu H. Expression of p53, epidermal growth factor receptor, Ki-67 and O6-methylguanine-DNA methyltransferase in human gliomas. Oncol Lett. (2013) 6:130–4. doi: 10.3892/ol.2013.1317
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Shen D, Liu T, Lin Q, Lu X, Wang Q, Lin F, et al. MGMT promoter methylation correlates with an overall survival benefit in Chinese high-grade glioblastoma patients treated with radiotherapy and alkylating agent-based chemotherapy: a single-institution study. PLoS ONE. (2014) 9:e107558. doi: 10.1371/journal.pone.0107558
PubMed Abstract | CrossRef Full Text | Google Scholar
34. The Cancer Genome Atlas Research Network (TCGA). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. (2008) 455:1061–8. doi: 10.1038/nature07385
CrossRef Full Text | Google Scholar
35. Benitez JA, Ma J, D’Antonio M, Boyer A, Camargo MF, Zanca C, et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat Commun. (2017) 8:15223. doi: 10.1038/ncomms15223
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. (2008) 455:1129–33. doi: 10.1038/nature07443
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Walker C, Baborie A, Crooks D, Wilkins S, Jenkinson MD. Biology, genetics and imaging of glial cell tumours. Br J Radiol. (2011) 84:S90–106. doi: 10.1259/bjr/23430927
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Gallia GL, Rand V, Siu IM, Eberhart CG, James CD, Marie SK, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. (2006) 4:709–14. doi: 10.1158/1541-7786.MCR-06-0172
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Zhao HF, Wang J, Shao W, Wu CP, Chen ZP, To ST, et al. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development. Mol Cancer. (2017) 16:100. doi: 10.1186/s12943-017-0670-3
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. (2016) 7:65696–06. doi: 10.18632/oncotarget.11602
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Cantanhede IG, de Oliveira JRM. PDGF family expression in glioblastoma multiforme: data compilation from Ivy glioblastoma atlas project database. Sci Rep. (2017) 7:15271. doi: 10.1038/s41598-017-15045-w
PubMed Abstract | CrossRef Full Text | Google Scholar
42. SongTao Q, Lei Y, Si G, YanQing D, HuiXia H, XueLin Z, et al. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. (2012) 103:269–73. doi: 10.1111/j.1349-7006.2011.02134.x
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. (2013) 13:345. doi: 10.1007/s11910-013-0345-4
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res. (2016) 22:1185–96. doi: 10.1158/1078-0432.CCR-15-1015
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. (1993) 53:2736–9.
PubMed Abstract | Google Scholar
46. Navis AC, van Lith SA, van Duijnhoven SM, de Pooter M, Yetkin-Arik B, Wesseling P, et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. (2015) 130:131–44. doi: 10.1007/s00401-015-1420-5
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Cruickshanks N, Zhang Y, Yuan F, Pahuski M, Gibert M, Abounader R. Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers. (2017) 9:E87. doi: 10.3390/cancers9070087
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Miyamoto M, Ojima H, Iwasaki M, Shimizu H, Kokubu A, Hiraoka N, et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer. (2011) 105:131–8. doi: 10.1038/bjc.2011.199
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Kwak Y, Kim SI, Park CK, Paek SH, Lee ST, Park SH. C-MET overexpression and amplification in gliomas. Int J Clin Exp Pathol. (2015) 8:14932–8.
PubMed Abstract | Google Scholar
50. Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. (2005) 7:436–51. doi: 10.1215/S1152851705000050
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. (2013) 369:1561–3. doi: 10.1056/NEJMcibr1309402
PubMed Abstract | CrossRef Full Text | Google Scholar
52. Plate KH, Breier G, Millauer B, Ullrich A, Risau W. Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. (1993) 53:5822–7.
PubMed Abstract | Google Scholar
53. Ma C, Li Y, Zhang X, Zhao G, Xu H. Levels of vascular endothelial growth factor and matrix metalloproteinase-9 proteins in patients with glioma. J Int Med Res. (2014) 42:198–204. doi: 10.1177/0300060513481924
PubMed Abstract | CrossRef Full Text | Google Scholar
54. Gravina GL, Mancini A, Colapietro A, Delle Monache S, Sferra R, Vitale F, et al. The small molecule ephrin receptor inhibitor, GLPG1790, reduces renewal capabilities of cancer stem cells, showing anti-tumour efficacy on preclinical glioblastoma models. Cancers. (2019) 11:E359. doi: 10.3390/cancers11030359
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Offenhäuser C, Al-Ejeh F, Puttick S, Ensbey KS, Bruce ZC, Jamieson PR, et al. EphA3 pay-loaded antibody therapeutics for the treatment of glioblastoma. Cancers. (2018) 10:E519. doi: 10.3390/cancers10120519
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Qazi MA, Vora P, Venugopal C, Adams J, Singh M, Hu A, et al. Cotargeting ephrin receptor tyrosine kinases A2 and A3 in cancer stem cells reduces growth of recurrent glioblastoma. Cancer Res. (2018) 78:5023–37. doi: 10.1158/0008-5472.CAN-18-0267
PubMed Abstract | CrossRef Full Text | Google Scholar
57. Halatsch ME, Gehrke EE, Vougioukas VI, Bötefür IC, A-Borhani F, Efferth T, et al. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J Neurosurg. (2004) 100:523–33. doi: 10.3171/jns.2004.100.3.0523
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PLoS ONE. (2012) 7:e44372. doi: 10.1371/journal.pone.0044372
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A, et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem. (2009) 284:7138–48. doi: 10.1074/jbc.M807111200
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Goodwin CR, Rath P, Oyinlade O, Lopez H, Mughal S, Xia S, et al. Crizotinib and erlotinib inhibits growth of c-Met+/EGFRvIII+ primary human glioblastoma xenografts. Clin Neurol Neurosurg. (2018) 171:26–33. doi: 10.1016/j.clineuro.2018.02.041
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. (2007) 6:1167–74. doi: 10.1158/1535-7163.MCT-06-0691
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. (2010) 12:95–103. doi: 10.1093/neuonc/nop015
PubMed Abstract | CrossRef Full Text | Google Scholar
63. Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. (2009) 27:579–84. doi: 10.1200/JCO.2008.18.9639
CrossRef Full Text | Google Scholar
64. Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. (2010) 12:1061–70. doi: 10.1093/neuonc/noq072
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Parker JJ, Dionne KR, Massarwa R, Klaassen M, Foreman NK, Niswander L, et al. Gefitinib selectively inhibits tumor cell migration in EGFR-amplified human glioblastoma. Neuro Oncol. (2013) 15:1048–57. doi: 10.1093/neuonc/not053
PubMed Abstract | CrossRef Full Text | Google Scholar
66. Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther. (2011) 10:1102–12. doi: 10.1158/1535-7163.MCT-11-0048
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. (2013) 85:1206–11. doi: 10.1016/j.ijrobp.2012.10.008
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group study N0074. Int J Radiat Oncol Biol Phys. (2011) 80:347–53. doi: 10.1016/j.ijrobp.2010.01.070
PubMed Abstract | CrossRef Full Text | Google Scholar
69. Eller JL, Longo SL, Kyle MM, Bassano D, Hicklin DJ, Canute GW. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. (2005) 56:155–62. doi: 10.1227/01.NEU.0000145865.25689.55
PubMed Abstract | CrossRef Full Text | Google Scholar
70. Eller JL, Longo SL, Hicklin DJ, Canute GW. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery. (2002) 51:1005–13; discussion: 1013–4. doi: 10.1227/00006123-200210000-00028
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. (2009) 20:1596–603. doi: 10.1093/annonc/mdp032
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. (1993) 362:841–4. doi: 10.1038/362841a0
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA. (1996) 93:14765–70. doi: 10.1073/pnas.93.25.14765
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Lee CG, Heijn M, di Tomaso E, Griffon-Etienne G, Ancukiewicz M, Koike C, et al. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. (2000) 60:5565–70.
75. Gossmann A, Helbich TH, Kuriyama N, Ostrowitzki S, Roberts TP, Shames DM, et al. Dynamic contrast-enhanced magnetic resonance imaging as a surrogate marker of tumor response to anti-angiogenic therapy in a xenograft model of glioblastoma multiforme. J Magn Reson Imaging. (2002) 15:233–240. doi: 10.1002/jmri.10072
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Yang SB, Gao KD, Jiang T, Cheng SJ, Li WB. Bevacizumab combined with chemotherapy for glioblastoma: a meta-analysis of randomized controlled trials. Oncotarget. (2017) 8:57337–44. doi: 10.18632/oncotarget.16924
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Kalpathy-Cramer J, Chandra V, Da X, Ou Y, Emblem KE, Muzikansky A, et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J Neurooncol. (2017) 131:603–10. doi: 10.1007/s11060-016-2332-5
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. (2010) 12:855–61. doi: 10.1093/neuonc/noq025
PubMed Abstract | CrossRef Full Text | Google Scholar
79. Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, et al. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. (2003) 63:7377–83.
PubMed Abstract | Google Scholar
80. Geng L, Shinohara ET, Kim D, Tan J, Osusky K, Shyr Y, et al. STI571 (Gleevec) improves tumor growth delay and survival in irradiated mouse models of glioblastoma. Int J Radiat Oncol Biol Phys. (2006) 64:263–71. doi: 10.1016/j.ijrobp.2005.08.025
PubMed Abstract | CrossRef Full Text | Google Scholar
81. Kilic T, Alberta JA, Zdunek PR, Acar M, Iannarelli P, O’Reilly T, et al. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res. (2000) 60:5143–50.
PubMed Abstract | Google Scholar
82. Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PMJ, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol. (2008) 26:4659–65. doi: 10.1200/JCO.2008.16.9235
PubMed Abstract | CrossRef Full Text | Google Scholar
83. Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. (2010) 96:393–402. doi: 10.1007/s11060-009-9976-3
PubMed Abstract | CrossRef Full Text | Google Scholar
84. de Boüard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. (2007) 9:412–23. doi: 10.1215/15228517-2007-024
CrossRef Full Text | Google Scholar
85. D’Amico R, Lei L, Kennedy BC, Sisti J, Ebiana V, Crisman C, et al. The addition of Sunitinib to radiation delays tumor growth in a murine model of glioblastoma. Neurol Res. (2012) 34:252–61. doi: 10.1179/1743132812Y.0000000005
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Grisanti S, Ferrari VD, Buglione M, Agazzi GM, Liserre R, Poliani L, et al. Second line treatment of recurrent glioblastoma with sunitinib: results of a phase II study and systematic review of literature. J Neurosurg Sci. (2019) 63:458–67. doi: 10.23736/S0390-5616.16.03874-1
PubMed Abstract | CrossRef Full Text | Google Scholar
87. Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL, et al. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer. (2010) 116:3663–9. doi: 10.1002/cncr.25275
PubMed Abstract | CrossRef Full Text | Google Scholar
88. Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett. (2011) 2:774–9. doi: 10.1021/ml200156t
PubMed Abstract | CrossRef Full Text | Google Scholar
89. Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, et al. Antitumor activity of NVP-BKM120-a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. (2012) 18:184–95. doi: 10.1158/1078-0432.CCR-11-1558
PubMed Abstract | CrossRef Full Text | Google Scholar
90. Wen PY, Touat M, Alexander BM, Mellinghoff IK, Ramkissoon S, McCluskey CS, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J Clin Oncol. (2019) 37:741–50. doi: 10.1200/JCO.18.01207
PubMed Abstract | CrossRef Full Text | Google Scholar
91. Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol. (2010) 12:559–69. doi: 10.1093/neuonc/nop058
PubMed Abstract | CrossRef Full Text | Google Scholar
92. Gwak HS, Shingu T, Chumbalkar V, Hwang YH, DeJournett R, Latha K, et al. Combined action of the dinuclear platinum compound BBR3610 with the PI3-K inhibitor PX-866 in glioblastoma. Int J Cancer. (2011) 128:787–96. doi: 10.1002/ijc.25394
PubMed Abstract | CrossRef Full Text | Google Scholar
93. Guessous F, Zhang Y, diPierro C, Marcinkiewicz L, Sarkaria J, Schiff D, et al. An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med Chem. (2010) 10:28–35. doi: 10.2174/1871520611009010028
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol. (2009) 4:69. doi: 10.1186/1748-717X-4-69
PubMed Abstract | CrossRef Full Text | Google Scholar
95. Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, et al. In vivo c-met pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Transl Oncol. (2013) 6:104–11. doi: 10.1593/tlo.13127
PubMed Abstract | CrossRef Full Text | Google Scholar
96. Li J, Di C, Mattox AK, Wu L, Adamson DC. The future role of personalized medicine in the treatment of glioblastoma multiforme. Pharmgenomics Pers Med. (2010) 3:111–27. doi: 10.2147/PGPM.S6852
PubMed Abstract | CrossRef Full Text | Google Scholar
97. Li J, Liang R, Song C, Xiang Y, Liu Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther. (2018) 11:731–42. doi: 10.2147/OTT.S155160
PubMed Abstract | CrossRef Full Text | Google Scholar
98. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. (2009) 16:748–54. doi: 10.1016/j.jocn.2008.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
99. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
PubMed Abstract | CrossRef Full Text | Google Scholar
100. Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. (2005) 353:2012–24. doi: 10.1056/NEJMoa051918
PubMed Abstract | CrossRef Full Text | Google Scholar
101. Li Q, Qiao G, Ma J, Li Y. Downregulation of VEGF expression attenuates malignant biological behavior of C6 glioma stem cells. Int J Oncol. (2014) 44:1581–8. doi: 10.3892/ijo.2014.2331
PubMed Abstract | CrossRef Full Text | Google Scholar
102. Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct. (2001) 26:25–35. doi: 10.1247/csf.26.25
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. (2009) 14:1131–8. doi: 10.1634/theoncologist.2009-0121
PubMed Abstract | CrossRef Full Text | Google Scholar
104. Latzer P, Shchyglo O, Hartl T, Matschke V, Schlegel U, Manahan-Vaughan D, et al. Blocking VEGF by bevacizumab compromises electrophysiological and morphological properties of hippocampal neurons. Front Cell Neurosci. (2019) 13:113. doi: 10.3389/fncel.2019.00113
PubMed Abstract | CrossRef Full Text | Google Scholar
105. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. (2007) 318:287–90. doi: 10.1126/science.1142946
PubMed Abstract | CrossRef Full Text | Google Scholar
106. Chakravarty D, Pedraza AM, Cotari J, Liu AH, Punko D, Kokroo A, et al. EGFR and PDGFRA co-expression and heterodimerization in glioblastoma tumor sphere lines. Sci Rep. (2017) 7:9043. doi: 10.1038/s41598-017-08940-9
PubMed Abstract | CrossRef Full Text | Google Scholar
107. Nazarenko I, Hede SM, He X, Hedrén A, Thompson J, Lindström MS, et al. PDGF and PDGF receptors in glioma. Ups J Med Sci. (2012) 117:99–112. doi: 10.3109/03009734.2012.665097
PubMed Abstract | CrossRef Full Text | Google Scholar
108. Frolov A, Evans IM, Li N, Sidlauskas K, Paliashvili K, Lockwood N, et al. Imatinib and Nilotinib increase glioblastoma cell invasion via Abl-independent stimulation of p130Cas and FAK signalling. Sci Rep. (2016) 6:27378. doi: 10.1038/srep27378
PubMed Abstract | CrossRef Full Text | Google Scholar
109. Pitz MW, Eisenhauer EA, MacNeil MV, Thiessen B, Easaw JC, Macdonald DR, et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol. (2015) 17:1270–4. doi: 10.1093/neuonc/nou365
PubMed Abstract | CrossRef Full Text | Google Scholar
110. Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. (2009) 115:140–8. doi: 10.1002/cncr.23972
PubMed Abstract | CrossRef Full Text | Google Scholar
111. Petterson SA, Dahlrot RH, Hermansen SK, K A, Munthe S, Gundesen MT, Wohlleben H, et al. High levels of c-Met is associated with poor prognosis in glioblastoma. J Neurooncol. (2015) 122:517–27. doi: 10.1007/s11060-015-1723-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Source:
Zuckerman Institute



{kind=link}