










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



The virus SARS coronavirus 2 (SARS-CoV-2) is the known cause of coronavirus disease 2019 (COVID-19). The “spike” or S protein facilitates viral entry into host cells.
Now a group of researchers from Lehigh and Seoul National University in South Korea and the University of Cambridge in the UK have worked together to produce the first open-source all-atom models of a full-length S protein.
The researchers say this is of particular importance because the S protein plays a central role in viral entry into cells, making it a main target for vaccine and antiviral drug development.
This video illustrates how to build the membrane system from their SARS-CoV-2 S protein models.
The model-building program is open access and can be found from the home page of CHARMM-GUI at the COVID-19 Archive.
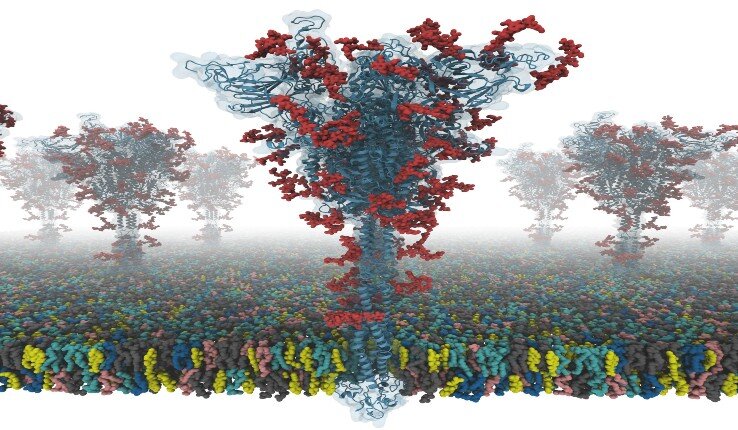
Developed by Wonpil Im, a professor in Lehigh’s Department of Biological Sciences and Bioengineering Department, CHARMM-GUI (graphical user interface) is a program that simulates complex biomolecular systems simply, precisely and quickly.
Im describes it as a “computational microscope” that enables scientists to understand molecular-level interactions that cannot be observed any other way.
“Our models are the first fully-glycosylated full-length SARS-CoV-2 spike (S) protein models that are available to other scientists,” says Im. “I was fortunate to collaborate with Dr. Chaok Seok from Seoul National University in Korea and Dr. Tristan Croll from University of Cambridge in the U.K.
Our team spent days and nights to build these models very carefully from the known cryo-EM structure portions. Modeling was very challenging because there were many regions where simple modeling failed to provide high-quality models.”
Scientists can use the models to conduct innovative and novel simulation research for the prevention and treatment of COVID-19, according to Im.
The S protein structure was determined with cryo-EM with the RBD up (PDB ID: 6VSB), and with the RBD down (PDB ID: 6VXX). But this model has many missing residues. So, they first modeled the missing amino acid residues, and then other missing domains.
In addition, they modeled all potential glycans (or carbohydrates) attached to the S protein. The glycans prevent antibody recognition, which makes it difficult to develop a vaccine. They also built a viral membrane system of an S protein for molecular dynamics simulation.
The team recommends reading the preprint paper, “Modeling and Simulation of a Fully-glycosylated Full-length SARS-CoV-2 Spike Protein in a Viral Membrane,” before using any of the models.
In a paper published May 26 in eLife, researchers found that the spike protein’s furin cleavage site is identical to a sequence in the human epithelial sodium channel, which likewise must be cut by furin in order to be activated.
The authors propose that the virus may be competing with the sodium channel for furin, and possibly disrupting its function, but that remains to be demonstrated.
“The paper’s really nice because it gets at this common view that many viruses co-opt parts of human cells to help them survive,” says David Perlin, who studies infectious diseases and is the chief scientific officer at Hackensack Meridian Health’s Center for Discovery and Innovation in New Jersey. He was not involved in the study.
This discovery came about when researchers at nference, an artificial intelligence company, started looking to see whether are there any sequences of amino acids in SARS-CoV-2 proteins that seemed surprising or unusual.
One that stuck out, says Venky Soundararajan, nference’s chief scientific officer, was a stretch of four amino acids present in the spike protein of 10,956 of 10,967 SARS-CoV-2 isolates from around the world, but not in the protein’s sequence in related coronaviruses, such as SARS-CoV or varieties that infect bats or pangolins.
Other groups had reported in February and April that this insertion forms a cleavage site for the human protease furin, which is thought to sever the two subunits of the spike protein to facilitate entry into human cells.
Soundararajan and colleagues strengthened this idea when they determined that the furin cleavage site is identical to a proven furin cleavage sequence in the alpha subunit of the human epithelial sodium channel, which plays a role in managing the balance of salt and fluid in many of the body’s cells.
This site is essential in the process of assembling the channel’s subunits into a functional whole capable of regulating sodium levels in a cell.
Next, the researchers turned to a platform they developed, which uses a database of single-cell RNA expression data from 65 previously published human and mouse studies, to look into gene expression of the sodium channel and other human genes known to be involved in SARS-CoV-2 infections.
They found that expression of the epithelial sodium channel’s gene overlaps with that of the gene for furin and for the primary SARS-CoV-2 receptor, ACE2, in the cell types most affected by the virus.
The coupling of ACE2, the epithelial sodium channel, and furin in the epithelial cells of the nasal cavity, the respiratory tract, and the gut supports the idea that those regions are the initial hubs of infection in the human body, Soundararajan says.
Perlin warns against making assumptions about ACE2 abundance based on transcript levels. “We have to always be a little careful going from gene expression to what’s assembled on the membrane,” he says.
Based on this RNA co-expression, the authors hypothesize that during a SARS-CoV-2 infection, the viral spike protein might compete with the human sodium channel for furin cleavage. If this competition disrupts the activation of the sodium channel, it could become dysregulated, which could interfere with its role in regulating fluid balance.
This could explain why COVID-19 patients sometimes end up with large amounts of fluid in the lungs.
The study “is all computationally based with no wet experiments. Hence we have zero information on what the authors are suggesting is at all correct,” cautions Vincent Racaniello, a virologist at Columbia University who was not involved in the work, in an email to The Scientist.
He also questioned the idea that the cleavage of the SARS-CoV-2 spike protein by human furin could usurp cleavage of the epithelial sodium channel.
It’s also not clear that this sodium channel is downregulated during viral infection. But if furin has a higher affinity for the virus and there is a high viral load in the same region where this sodium channel is expressed, “there is potential there that you’d have less processing of that alpha subunit of the sodium channel and this potentially could also [affect] sodium channel function locally,” says Perlin. “Is that what’s happening? I’m not sure, but it’s an interesting hypothesis.”
“Most computational analysis requires some experimental validation,” says Dario Ghersi, a computational biologist at the University of Nebraska Omaha who did not participate in the study.
That this recognition site is present in the majority of SARS-CoV-2 isolates implies that it is important to the virus, meaning the sequence could be of interest for vaccine and therapeutic development. “What they found seems pretty solid to me,” he adds.
P. Anand et al., “SARS-CoV-2 strategically mimics proteolytic activation of human ENaC,” eLife, doi:10.7554/eLife.58603, 2020.
More information: Hyeonuk Woo et al. Modeling and Simulation of a Fully-glycosylated Full-length SARS-CoV-2 Spike Protein in a Viral Membrane (2020). DOI: 10.1101/2020.05.20.103325



{kind=link}
[…] COVID-19 : Researchers produced the first open-source all-atom models of a… […]