









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



After only a few days caring for critically ill COVID-19 patients at the start of the outbreak in New York City, Aakriti Gupta, MD, realized that this was much more than a respiratory disease.
“I was on the front lines right from the beginning. I observed that patients were clotting a lot, they had high blood sugars even if they did not have diabetes, and many were experiencing injury to their hearts and kidneys,” says Gupta, one of the first Columbia cardiology fellows to be deployed to the COVID intensive care units at Columbia University Irving Medical Center.
In early March, there wasn’t much clinical guidance on the non-respiratory effects of COVID-19, so Gupta decided to coalesce findings from studies that were just beginning to appear in the literature with what the physicians were learning from experience.
Gupta, along with senior author Donald Landry, MD, Ph.D., chair of medicine at Columbia University Vagelos College of Physicians and Surgeons, organized senior co-authors, and Gupta, along with two other colleagues, Mahesh Madhavan, MD, a cardiology fellow at CUIMC, and Kartik Sehgal, MD, a hematology/oncology fellow at Beth Israel Deaconess Medical Center/Harvard Medical School, mobilized clinicians at Columbia, Harvard, Yale, and Mount Sinai Hospital, among other institutions, to review the latest findings on COVID-19’s effect on organ systems outside the lungs and provide clinical guidance for physicians.
Their review – the first extensive review of COVID-19’s effects on all affected organs outside the lungs – was published in Nature Medicine.
“Physicians need to think of COVID-19 as a multisystem disease,” Gupta says. “There’s a lot of news about clotting but it’s also important to understand that a substantial proportion of these patients suffer kidney, heart, and brain damage, and physicians need to treat those conditions along with the respiratory disease.”
Blood Clots, Inflammation, and an Immune System in Overdrive
“In just the first few weeks of the pandemic, we were seeing a lot of thrombotic complications, more than what we would have anticipated from experience with other viral illnesses,” says Sehgal, “and they can have profound consequences on the patient.”
Scientists think these clotting complications may stem from the virus’s attack on cells that line the blood vessels. When the virus attacks blood vessel cells, inflammation increases, and blood begins to form clots, big and small.
These blood clots can travel all over the body and wreak havoc on organs, perpetuating a vicious cycle of thromboinflammation.
To combat clotting and its damaging effects, clinicians at Columbia, many of whom are co-authors on this review, are conducting a randomized clinical trial to investigate the optimal dose and timing of anticoagulation drugs in critically ill patients with COVID-19.
The untempered inflammation can also overstimulate the immune system, and though doctors initially shied away from using steroids to globally suppress the immune system, a recent clinical trial has found that at least one steroid, dexamethasone, reduced deaths in ventilated patients by one-third.
Randomized clinical trials are underway to target specific components of thromboinflammation and the immune system, such as interleukin-6 signaling.
“Scientists all over the world are working at an unprecedented rate towards understanding how this virus specifically hijacks the normally protective biological mechanisms. We hope that this would help in the development of more effective, precise, and safer treatments for COVID-19 in the near future,” says Sehgal.
Straight to the Heart
Clots can cause heart attacks, but the virus attacks the heart in other ways, one author says.
“The mechanism of heart damage is currently unclear, as the virus has not been frequently isolated from the heart tissue in autopsy cases,” says Gupta.
The heart muscle may be damaged by systemic inflammation and the accompanying cytokine release, a flood of immune cells that normally clears up infected cells but can spiral out of control in severe COVID-19 cases.
Despite the degree of heart damage, physicians were not able to utilize the diagnostic and therapeutic strategies, including heart biopsies and cardiac catheterizations, that they would normally use during the early stages of the pandemic given the need to protect personnel and patients from viral transmission. This has changed as the disease prevalence has gone down in New York CIty.
Kidney Failure
Another surprising finding was the high proportion of COVID-19 patients in the ICU with acute kidney damage.
The ACE2 receptor used by the virus to gain entry into the cells is found in high concentrations in the kidney and could likely be responsible for the renal damage. Studies in China reported renal complications, but in New York City, clinicians saw renal failure in up to 50% of patients in the ICU.
“About 5 to 10% of patients needed dialysis. That’s a very high number,” Gupta says.
Data regarding long-term renal damage are currently lacking, but a significant proportion of patients will likely go on to require permanent dialysis.
“Future studies following patients who experienced complications during hospitalizations for COVID-19 will be crucial,” notes Madhavan.
Neurological Effects
Neurological symptoms, including headache, dizziness, fatigue, and loss of smell, may occur in about a third of patients.
More concerning, strokes caused by blood clots occur in up to 6% of severe cases and delirium in 8% to 9%.
“COVID-19 patients can be intubated for two to three weeks; a quarter require ventilators for 30 or more days,” Gupta says.
“These are very prolonged intubations, and patients need a lot of sedation. ‘ICU delirium’ was a well known condition before COVID, and the hallucinations may be less an effect of the virus and more an effect of the prolonged sedation.”
“This virus is unusual and it’s hard not to take a step back and not be impressed by how many manifestations it has on the human body,” says Madhavan.
“Despite subspecialty training as internists, it’s our job to keep all organ systems in mind when caring for the patients in front of us. We hope that our review, observations, and recommendations can help other clinicians where cases are now surging.”
The paper is titled, “Extrapulmonary manifestations of COVID-19.”
The Phylogenetic homology and its correlation with the possibilities of developing isolated vasculopathy in SARS-CoV-2 infected patients
Coronaviruses are single stranded ( +) RNA viruses that appear like a crown shape under an electron microscope due to presence of spike glycoproteins on the outer coat.
The Coronaviridae family of order Nidovirales can be classified into four genera of coronaviruses like alpha-coronavirus (α-CoV), beta-coronavirus (β-CoV), delta-coronavirus (δ-CoV), and gamma-coronavirus (γ-CoV) [2].
The divergence of Coronviruses into their subgroups is estimated to have occurred 5000 years ago [3]. The members of this virus group can cause respiratory, enteric, hepatic, and neurological diseases in different animal species, including camels, cattle, cats, and bats. Till date, seven Human-CoVs (HCoV) – capable of infecting humans have been identified (https://www.ncbi.nlm.nih.gov/books/NBK554776).
The genome of the new HCoV, isolated from a patient with atypical pneumonia after visiting Wuhan, had 89% nucleotide identity with bat SARS-like-CoVZXC21 and 82% with that of human SARS-CoV [4]. The size of its single-stranded RNA genome vary from 26 to 32kbs.
Although its origins are not entirely understood, these genomic analyses suggest that SARS-CoV-2 probably evolved from a strain found in bats. However, identification of a potential intermediate mammalian host connecting bats and humans, remains incomplete [1].
Vasculopathy: a phylogenetic ‘footprint’ manifestation in SARS?
Systemic cytokine activation with reactive hemophagocytic syndrome, acute tubular necrosis, skeletal muscle fiber necrosis and lymphoid depletion in spleen, observed SARS-CoV patients are reminiscent of those reported for fatal influenza virus subtype H5N1 disease in 1997 [5].
SARS patients also exhibit gastrointestinal symptoms [6] along with splenic atrophy and lymphadenopathy [7]. Diarrhoea is a very frequent manifestation among SARS patients (48.6% of recruited patients); therefore a possible pattern in gastrointestinal vasculature involvement at early point of the infection is clear [6].
Prominent findings based on autopsies of SARS-CoV patients depict that SARS-CoV infection is a systemic vascular disease with widespread extrapulmonary dissemination among various organ systems, being evidential in the form of viral shedding in respiratory secretions, stools, urine, and even sweat [8, 9].
MERS which shares 50% phylogenetic homology to SARS-CoV-2 [10] occurred during 2012 mainly in Arab and Middle East, also has similar tissue tropism but differs in the attachment receptor. The disease presents as severe respiratory infection often with shock, acute kidney injury (AKI), and coagulopathy [11].
The receptor for MERS-CoV attachment identified as dipeptidyl peptidase-4 (DPP4/CD26) [12], is expressed in lung, liver, placenta, skeletal muscle, heart, brain, pancreas [13]. The presence of this receptor may be linked to the susceptibility of visceral vasculature including the brain, to infection [14]; which needs further study.
Patient reports suggest that MERS-CoV infection targets multiple systems including pulmonary, cardiovascular, renal, coagulation, gastrointestinal tract, muscular. Angiotensin converting enzyme 2 (ACE2) being the primary attachment receptor for SARS-CoV-2, is a single-pass transmembrane protein with protease activity that cleaves the vasoconstrictor angiotensin II into the vasoprotective substance, angiotensin 1–7 [15, 16].
In humans, a broad expression of ACE2 receptor has been found in heart, kidney, gastrointestinal tract, and in other tissues; potentially explaining the multi-organ injury observed with SARS-CoV-2 infection [17].
Immuno-phylogenetic homology in vasculopathy between SARS-CoV and SARS-CoV2
Cytopathic viruses, including SARS-CoV-2 [18], induce injury and death of virus-infected cells and tissues as part of the viral replicative cycle in airway epithelial cells [19].
Pyroptosis, a highly inflammatory form of programmed cell death commonly seen with cytopathic viral infection,has been found to be associated with subsequent vascular leakage, in patients with SARS-CoV [20, 21].
According to recent study it has been found that SARS-CoV Viroporin 3a triggered the activation of the nod like receptor protein-3(NLRP3) inflammasome along with the secretion of IL-1β in bone marrow-derived macrophages, leading to SARS-CoV induced cell pyroptosis [21].
Interestingly, patients infected with SARS-CoV-2 have been found with elevated level of IL-1β in the serum [22]. IL-1β, a downstream indicator of cell pyroptosis, can potentiate cell pyroptotic cascade in lymphocytes, macrophages and vascular endothelium, leading to vasculopathy in COVID-19 patients [Fig. 1].
Moreover, as both classical and non-classical pyroptosis signalling can induce the release of IL-1β, it remains unclear as to which pathway is involved in COVID-19 [23].
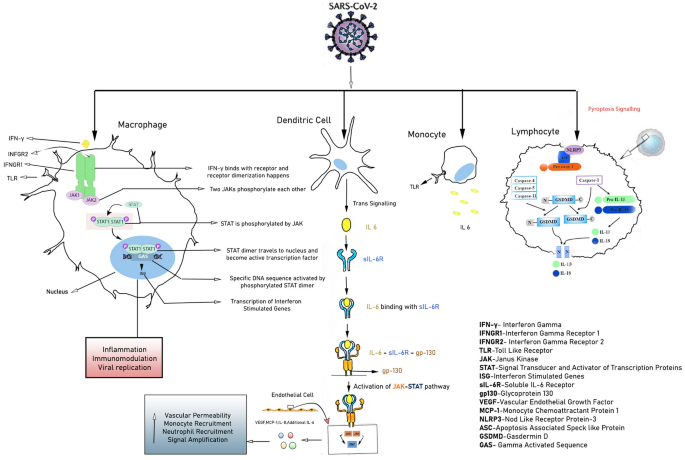
Immunological interplay can augment SARS-CoV-2 mediated epithelial and endothelial injury
Immunological response of COVID-19 infected patients exhibits dynamic variations based on the magnitude of disease severity. Initially upon viral invasion, activation of innate immune system leads to the accumulation of neutrophils which is found to be higher among critically ill patients in comparison to the patients with mild to moderate degree of infection [24].
Collective evidences reflect that first line defence of COVID-19 infection includes an elevation of several cytokines such as IL-1β, IL-1RA,IL-7,IL-8,IL-9,IL-10,IL-6, fibroblast growth factor(FGF),granulocyte macrophage colony stimulating factor (GM-CSF), interferon gamma (IFN- γ),granulocyte colony stimulating factor (G-CSF), platelet derived growth factor (PDGF), tumour necrosis factor (TNF- α),vascular endothelial growth factor (VEGF) as well as some chemokines such as interferon-gamma-inducible-protein (IP-10), monocyte chemoattractant protein-1 (MCP1), macrophage inflammatory protein 1 alpha (MIP1α) [22, 25].
It is important to mention that higher amount of IL-2, IL-6, IL-7, IL-10, G-CSF,IP-10,MCP-1,MIP-1α, and TNF-α are detected among severe COVID-19 patients (ICU patients) rather than the non-ICU patients [22, 24, 25] [Fig. 2].
Henceforth, it can be inferred that ‘cytokine storm’ or ‘hypercytokinemia’ is one of the significant pathophysiological mechanisms of COVID-19, as previously found in SARS and MERS [26].
Accumulative evidences suggest that IL-6 plays a vicious role behind the cytokine storm and subsequent endothelial injury as illustrated in Fig. 1 [27]. Highly inflammatory monocyte derived ficolin-1 (FCN1+) macrophage population in the broncho-alveolar-lavage (BAL) fluid of critically ill COVID-19 patients is related with the secretion of inflammatory cytokines including IP-10, MCP-1, MIP-1α (Clinical Trials.gov. Identifier NCT04273321, Efficacy and Safety of Corticosteroids in COVID-19. 2020 Feb 24; Available from: https://clinicaltrials.gov/ct2/show/NCT04273321). Adaptive immune responses among COVID-19 infected patients have been detected within an average period of one week of onset of symptoms (https://bit.ly/2zpaZ1a).
Autopsy of a COVID-19 patient revealed increased number of T-lymphocytes in peripheral as well as visceral vasculature, mostly in lungs [28]. Hence, this report indicates that T cells are accumulated at the major site of viral infection in order to control it.
Besides, it is also important to note, that depletion of CD8+TC cells and NK cells is noticeably more in COVID-19 patients unlike mild to moderate group which establishes another important diagnostic phenomenon of this viral infection; i.e. lymphocytopenia [26].
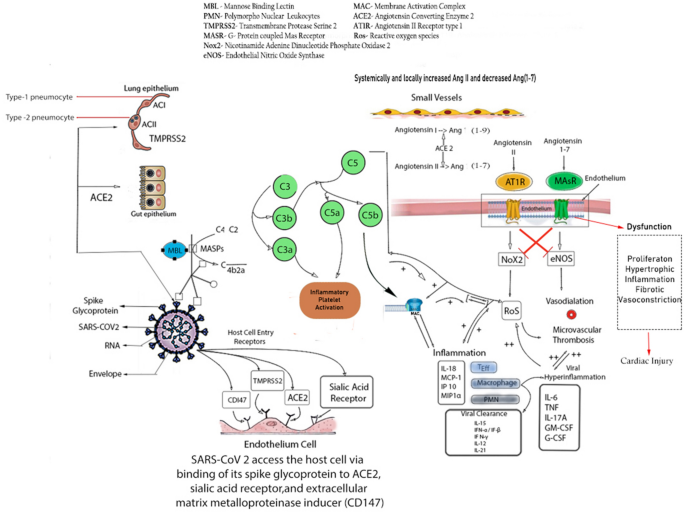
SARS-CoV-2 enters the human host primarily by binding with ACE2 receptors, a key protein associated with both adaptive and innate immune response, B cell activation and ‘Cytokine Storm’. Interestingly a number of literatures suggest that higher expression of ACE2 also enhances viral replication cycle after the entry into the host cells which in turn mediates the alteration of epithelial transcriptome.
ACE2 expression also leads to the activation of neutrophils, NK-cells, Th-17cells, Th-2 cells, Th-1cells, dendritic-cells, TNF-α secreting cells and elaboration of several proinflammatory cytokines [29].
According to a recent single cell-RNA sequencing(scRNAseq) dataset based study, SARS-CoV-2 can exploit human or species specific interferon driven upregulation of ACE2 gene and a subsequent enhancement of ACE2 receptor surface expression in type-II pneumocytes, illeal absorptive enterocytes and nasal goblet cells [30]; causing a strategic acceleration of the infection.
Considering a significant endothelial ACE2 expression across different visceral vasculature [31], and pleiotropic response of type-1 IFN (IFN-α) toon endothelial cells [32], which may affect the endothelial integrity and subsequent injury [33]; a deeper understanding of the vascular response upon SARS-CoV-2 infection is important.
Additionally interferon upon binding with its receptor induces phosphorylation of transcription factors such as STATs by JAK and relocalization of these transcription factors inside the nucleus activates interferon stimulated genes (ISG).
These ISGs are asoociated with inflammation, immunomodulation and interfere viral replication [30, 34, 35]. Furthermore, increased amount of Angiotensin-II (Ang-II) impair the level of protective Ang (1–7).
Besides Ang-II binds to Angiotensin-II receptor type-1 (AT1R),and thereby subsequently enhances the synthesis of downstream nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) and reactive oxygen species (ROS).Ang (1–7), on the other hand binds to G-protein coupled Mas receptor (MasR) and a subsequent overexpression of a potential vasodilator i.e. endothelial nitric oxide synthase (eNOS) is evident.
A mutual co-supression of individual downstream pathway between MasR and AT1R plays a pivotal role towards endothelial damage, thrombosis inflammation and subsequent vasculopathy [36].
Additionally, as previously mentioned, aggravated inflammatory response has been observed among the infected individuals which may further lead to Pyroptosis, specially observed in lymphocytes, through the activation of NLRP inflammasome.
The crosstalk involved in the activation of the signalling between NLRP3m, IL-1β,IL-18 and gasdermin-D (GSDMD) as illustrated in Fig. 1, needs further study in COVID-19 patients [20]. In case of SARS-CoV-2 infection, lectin pathway (LP) mostly generates inflammatory response.
In this pathway, binding of mannose binding lectin (MBL) to mannose and N-acetylglucosamine (NAG) residues in the virus envelop or virus-infected cells activate MBL-associated serine protease 2 (MASP-2) [37, 38]. MASP-2 is considered as a key regulator of LP which results in an activation of complement system and inflammatory lung injury [39].
Furthermore, IHC staining of fixed lung tissue samples from dead COVID-19 patients revealed the presence of MBL,MASP-2,C4α,C3, C5b deposition in type-I and type-II alveolar epithelial cells along with some hyperplastic pneumocytes, exudates in alveolar space with necrotic cell debris and inflammatory cells [39] [Fig. 2].
SARS-CoV-2 mediated alteration in haematological parameters can reflect underlying vascular endothelial damage and coagulopathy
SARS-CoV-2 mediated alteration in human heme metabolism
Recent studies have revealed a massive demand of porphyrins for viruses to survive. As a consequence, SARS-CoV-2 displays an unusual binding affinity towards haemoglobin.
According to this study, E2 glycoprotein, envelope protein, nucleocapsid phosphoprotein, orf1ab, ORF7a and ORF8 of the virus could bind to porphyrin of the 1-β chain of haemoglobin (Hb) to form a complex by displacing Fe+2 from heme residues [40].
Henceforth such unusual attack can lead Hb to carry less amount of oxygen and carbon di-oxide, worsening the situation of patients already with a severe respiratory distress.
SARS-CoV2 can augment intravascular haemolysis and hypoxia
Inhibition of the human heme synthesis, can also trigger the onset of haemolysis [41]. Considering a recent finding that people with blood group-O are not easily infected by SARS-CoV-2, whereas those with blood group A have a significantly higher risk of acquiring the infection [42], it can be speculated that immune haemolysis may be the major mechanism of viral protein mediated attack on Hb.
Therefore, SARS-CoV-2 mediated systemic intravascular haemolysis as well as alteration in heme metabolism can lead to iron overload, oxidative stress, additional respiratory distress, and hypoxia.
This leads to a cascade of immune response; with increased TNF-α, IL-6, IL-1, ICAM-1 levels, in pulmonary and other visceral microvasculature with subsequent capillary endothelial injury [43].
In conjugation to the pro-coagulant effectors derived from pro-inflammatory cytokines, neutrophilic extra cellular traps(NETS) and polyphosphates, the usual thrombo-protective state of small to micro-vascular endothelial cells is dissociated; leading to the development of microvascular thrombosis [44].
It has also been documented that eosinophil count can be a significant marker for COVID-19 prognosis [45, 46]. The role of eosinophils and IL-5 in COVID-19 and its impact in visceral microvasculature especially in pulmonary vasculature, needs to be further investigated [47].
COVID-19 mediated complement activation and neutrophilia
Literatures on post-mortem histopathological studies clarify evidences of vascular deposition of C5b-9 as a key feature of many micro thrombotic syndromes, regardless of the particular syndromic complex, including catastrophic antiphospholipid antibody syndrome, atypical haemolytic uremic syndrome(a-HUS), purpura fulminans and severe multi-organ malignant atrophic papulosis, and can be responsive to anti-complement therapies [48,49,50,51,52].
Tissue neutrophilia may be attributable to the neutrophil chemoattractant properties of complement. Both neutrophil and complement are key sentinels of innate immunity and also modulate thrombogenic pathways, the latter thought related to C5a receptor/tissue factor cross-talk mediated by neutrophils [53].
Neutrophilia in COVID-19 positive patients is correlated with a poor prognosis and could be an index of the extent of complement activation [54, 55]. Another finding suggests an increase in peripheral blood neutrophil vacuolization and granule content in patients is consistent with an activated state, even if absolute numbers are not elevated [36].
Henceforth, neutrophil count could be important marker for underlying systemic vascular endothelial as well respiratory epithelial cells and endothelial damage associated micro-vascular pro-thrombotic changes, which may eventually lead to Multiple organ dysfunction (MOD) worsening the overall prognosis.
Alteration in coagulation factors
Besides SARS-CoV2 infection is also associated with increased coagulation abnormalities including increased fibrinogen and higher level of D-dimers, that have been associated with higher morbidities and mortality [56, 57].
Interestingly, a correlation of IL-6 and fibrinogen levels has been reported, demonstrating and confirming the link between inflammatory and pro-coagulopathic changes [44].
Evidences also suggest, co-manifestation of sepsis induced coagulopathy (SIC) and disseminated intravascular coagulopathy (DIC) especially in severe diseases as well as in non-survivors along with a report of hyper-fibrinolytic consumptive DIC with a bleeding diathesis [58, 59].
Recent publications and pre-print reports also suggest incidences of venous thrombo-embolism events (VTE) in COVID19 positive critically ill patients admitted to ICU [60]. Henceforth a regular Monitoring of fibrinogen, Prothombin time (PT), activated partial thromboplastin time (aPTT), D-dimer, and renal function should be performed along with a subsequent risk adapted modulation in anti-coagulant therapy.
Besides, in a recent control cohort study of 540 specimens, the percentage positivity of lupus anticoagulant was significantly higher in patients with COVID-19 than in control group. Most of these patients admitted with a prolonged aPTT were positive for lupus anticoagulant and an associated factor XII deficiency [61]. Hence a further study is required to elucidate the role if any, of lupus anticoagulant in the pathogenesis of SARS-CoV-2 mediated thrombosis.
Impact of SARS-CoV-2 in cardio-pulmonary microvasculature
Derrangment of cardiac-microvasculature in SARS-CoV-2
To elucidate the topography of ACE2 in the heart a single nuclear transcriptome analysis was performed on an isolated myocardial nucleus which showed the maximum proportion of ACE2 receptors in the pericytes.
The expression in pericytes of human heart makes it vulnerable to SARS-CoV-2 infection [62]. This microvascular injury especially in the coronary microvasculature may attribute to the development of cardiomyopathy in debilitating patients.
The inter communication between pericytes and endothelial cells (EC) via the angiopoietin ligands (ANGPT1/2) with tyrosine kinase with immunoglobulin like and EGF like domains (TIE2) receptor interaction is vital for the EC survival and suitable microvascular environment [63]. Viral invasion of pericytes leads to disruption of this ambiance leading to micro-vascular injury.
SARS-CoV-2 can affect the heart by an array of mechanisms which ranges from myocardial insult due to direct cardiomyocyte invasion [64] to oxidative stress as a sequela of hypoxia and increased myocardial oxygen demand.
A case report of a 47-year-old afro Caribbean with past history of myopericarditis presenting with dry cough, breathlessness, chest pain was later diagnosed with cardiac tamponade evident on echocardiogram and elevated troponin-T levels [65].
Therapeutic pericardiocentesis was done which relieved the symptoms. The subject tested positive for COVID-19. Complications like fulminant myocarditis in genetically confirmed COVID-19 on sputum testing has also been reported.
A 63-year old man with COVID-19 presenting with cardiac symptoms, lab panel revealed elevated IL-6 levels (272 pg/ml) and trop-I levels with a reduced left ventricular ejection fraction on echocardiography. All other viral aetiologies of myocarditis were ruled out. symptoms improved on receiving antiviral therapy and mechanical life-support [66].
A case series report shows elevated ST-segments on electrocardiography in a set of 18 confirmed COVID-19 patients with no evidence of pre-existing heart disease indicating development of myocardial infarction.
Obstruction in the coronary circulation had precipitated in a subset of patients which was revealed through angiography. Elevated D-dimer levels were persistent in all of the cases [67]. After analysing the above reports, consideration of SARS-CoV-2 as an inciting factor in development of a myocardial pathology should be pondered upon.
Another report of autopsies performed on four confirmed COVID-19 deceased could be in favour of above cardiac involvement. In all the cases the clinical course escalated from an episode of mild fever, cough into full blown acute respiratory distress syndrome with respiratory decompensation.
Past history of obesity and hypertension, controlled on medication, was present in all of them with type-2 diabetes in three of them, chronic kidney disease in two of them. Mechanical ventilation and antimicrobial or steroid therapy (dexamethasone) were to no avail.
Laboratory reports revealed elevated PT, aPTT, fibrinogen degradation products (FDP), FIB and D-dimer levels, consistent with a prothrombotic state along with neutrophilia and lymphocytopenia.
Subsequent cardiac examination on three patients revealed cardiomegaly and right ventricular dilatation. Despite of the absence of confluent myonecrosis or adjacent lymphocytic infiltrate, individual cell necrosis was present in all of them on histopathological examination [68].
As per the Chinese CDC the co morbidities associated with increased COVID-19 deaths were refractory hypertension, diabetes mellitus, age greater than 70 and pre-existing heart diseases [69].
A retrospective data analysis evaluated over 150 lab confirmed patients of SARS-CoV-2 revealed that cardiovascular diseases were associated with significantly higher chances of death [70].
According to another retrospective cohort study based on patients admitted in two hospitals of Wuhan, elevated levels of IL-6, troponin I, lactate dehydrogenase was a frequent finding. Autopsy on a 53-year-old female with chronic kidney disease revealed acute myocardial infarction.
Old age, higher SOFA-score (sequential organ failure assessment), elevated D-dimer levels (> 1 µg/ml) were associated with a poor prognosis [56]. Derangement in coagulation profile is persistent in all SARS-CoV-2 cases. This leads to visceral microvascular damage in the form of endothelial injury and thrombosis.
In a prospective study among 94 confirmed cases of COVID-19 in Renmin hospital of Wuhan University, coagulation profile was monitored over a period of time and compared with 40 healthy controls. Results showed reduced antithrombin levels and increased PT, aPTT, FDP and D-dimer levels in COVID-19 patients compared to healthy subjects [71].
Impact of SARS-CoV-2 in pulmonary microvasculature: do we need to revise the tag of ARDS?
Up-till now, respiratory symptoms are the primary manifestations in COVID-19 patients by far. Lungs seem to be the port of entry widely used by the virus resulting in acute respiratory distress in most of the patients. But, the pattern of lung injury is not typical of classic Acute Respiratory Distress Syndrome (ARDS) in COVID-19 patients with the absence of hallmark signs like diffuse alveolar damage (DAD) with hyaline membrane formation and type-II pneumocyte hyperplasia [36].
Moreover, it has been observed that the lung mechanics is not much hindered in the process, evident on preserved lung compliance and high shunt fraction [36].
Study reveals the lung injury in these diseases is associated with membrane attack complex (c5b-9) deposition in the pulmonary microvasculature along with c4d and MASP2, inclining towards a potential complement mediated vascular injury. Purpuric skin lessons were also present in these patients which revealed a pauci-inflammatory thrombogenic vasculopathy characterised by similar complement deposition in vessel wall [36].
Coagulative vasculopathy may be the basis of visceral damage triggered by complement mediated endothelial injury. Monoclonal antibodies against complement factors like eculizumab (targeted against C5), narsoplimab (lectin-pathway-inhibitor by targeting MASP-2) could be considered for therapy. Another report supporting the above mechanism shows fibrin deposition in the alveolar capillaries consistent with a coagulopathy related damage to the pulmonary bed [68].
The alveolar capillaries revealed thrombi formation and surrounding edema with scattered hyaline membrane formation. Another noticeable finding was presence of CD-61 + megakaryocytes, which could be the source of platelet production subsequently leading to aggregation [68].
Besides, studies have shown that immunostaining of lung specimen with antibody to Rp3 NP protein of SARS-CoV-2 shows increased expression in lung epithelia [72].
A summary of two cases of COVID-19 illustrates the development of acute pulmonary embolism evident on CT pulmonary angiogram in both of them. Blood profile showed elevated d-dimer levels persistent with acute pulmonary embolism [73].
Autopsy on four COVID-19 infected cases with pre-existing diseases (chronic lymphocytic leukemia, renal transplant, cirrhosis, hypertension, diabetes) revealed type-2 pneumocyte hyperplasia, hyaline membrane formation, fibrin clusters in airspaces [74]. Above facts depict that COVID-19 may be associated with a hypercoagulable state leading visceral thrombosis and subsequent damage.
Involvement of Renal microvasculature in SARS-CoV-2
COVID-19 patients can develop Acute Kidney Injury (AKI) as a complication. Incidence of AKI in COVID-19 patients ranges from 0.5 to 28%. Previous data has shown the incidence of 0.5 percent, 5.1 percent, 15 percent and 28 percent in four different studies [75,76,77]. A study based in Wuhan, China suggested AKI to be uncommon in COVID-19 as they reported patients returning to normal after follow up and temporary abnormal renal function is due to hypoxemia [78].
AKI is considered as a critical prognostic factor for COVID-19 and is curable by interventions. Endotoxins, sepsis, rhabdomyolysis, cytokine storm leads to acute tubular necrosis which subsequently causes AKI [79, 80].
Other possible ways of renal damage [79] are renal medullary hypoxia due to alveolar damage, rhabdomyolysis leading to tubular toxicity, cardiorenal syndrome (in patients of cardiomyopathy/viral myocarditis), endothelial damage, high peak airway pressure and intrabdominal hypertension leading to renal compartment syndrome, third space fluid loss and hypotension leading to renal hypoperfusion. In case reports on COVID-19, rhabdomyolysis has been reported on presentation [81] and as a complication [82].
There have been studies indicating the direct cytopathic effect on kidneys of COVID-19 [83, 84]. SARS-CoV-2 enters into cells via host ACE2 receptors. According to a PubMed Database, RNA expression of ACE2 is nearly 100 fold more in GI, kidney and urinary tract than in Respiratory tract [Gene ID: 59,272,Primary Source-HGNC:HGNC:13557]. Another preprint indicates that ACE2 and TMPRSSs family [host cell receptor and the key protease for COVID-19] are significantly co expressed in podocytes and Proximal Convoluted Tubule [85].
Renal histopathological analysis of 26 autopsies of patients with COVID-19 showed clusters of coronavirus particles with spikes in the tubular epithelium and podocytes. ACE2 was upregulated and immunostaining with SARS-CoV nucleoprotein Ab was positive in tubules [83]. Fragments of coronavirus in patients with COVID-19 have been found in urine [86].
This raises the possibility of virus particles shedding in the urine and being a possible source of infection. Mortality risk for COVID-19 patients with AKI was significantly higher than without AKI [Hazard Ratio HR 5.3] [77]. Multiple studies have suggested increase in blood urea nitrogen (BUN), creatinine, proteinuria, haematuria as independent risk factors for mortality in COVID-19 [76].
For SARS- CoV infection, Uraemia is associated with extensive impairment of lymphocyte and granulocyte function and T cells play a key role in recovery [87]. Due to similarities of SARS-CoV-2 structure with SARS-CoV, it could raise a potential concern for haemodialysis patients infected with SARS-CoV-2 [88].
Haemodialysis patients usually have multiple comorbidities and frequent visits to dialysis centre put them under risk of infection. Guidelines and management plans for patients with COVID-19 undergoing dialysis are very important.
Neuro-vasculopathy in SARS-CoV-2
From the perspective of neurological manifestations of COVID-19, evidences gathered in favour of vasculopathy thus far principally correspond to cerebro-vascular events (CVEs).
After the early study from Wuhan [89] that documented the frequency of neurological consequences of SARS-CoV-2 infection, a subsequent single-centre study from the same province of China reported that more than 5% of such manifestations can be attributed to occurrence of stroke, ischemic much more than hemorrhagic [90].
Reports of some isolated cases across the world have reiterated the notion that thrombotic events are indeed more likely. For instance, a recent case study from USA has documented occurrence of large vessel occlusion involving left middle cerebral artery in a confirmed case of SARS-CoV-2 infection [91].
Notably, the patient initially manifested with STEMI for which he was put on treatment including anti-coagulant therapy. Arterial Doppler of lower limbs, which was performed because he had suggestive symptoms, revealed occlusion in multiple arterial segments on both sides.
Small vessel lacunar infarct in left middle cerebral artery territory in the background of paroxysmal atrial fibrillation has been documented from Hubei province in China [92]. Similarly, a case described from a different province (Xi’an) in China records the co-occurrence of right middle cerebral artery infarction and deep vein thrombosis in both lower limbs [93].
As opposed to the previous two reports where thrombotic manifestations preceded SARS-CoV-2 detection, the latter patient was a confirmed case of severe COVID-19 and developed thrombotic events around 2 weeks into the illness. However, all three cases have been documented to affected elderly patients in their 8th decade of life and with co-morbid conditions.
In recent times, it has been speculated that the virus, so far considered as a primary lung pathogen, may indeed possess some vascular invasive potential resulting in widespread thrombotic manifestations including CVEs. The idea of ‘accelerated thrombosis’, as in other disorders of immune dysregulation seems more realistic with the passage of time.
This observation is important because of two reasons. First, the diagnostic challenges become even steeper with these atypical presentations. Second, therapeutic armamentarium faces the need for an up gradation, with possible inclusion of anti-coagulants.
Noteworthy, the size of cerebral vessels seemingly predisposed to viral invasion include all three varieties, namely large, medium and small. This observation however is in need of substantiation with larger studies, for it should be accommodated in the therapeutic planning of such patients.
Arguments have also been framed about the putative role of ACE2 receptor tropism of the viral particle in causing cerebral hemorrhage. In accordance with the initial Wuhan report [90], while ischemic strokes in COVID-19 have been documented more often, hemorrhagic strokes have rarely surfaced up.
A recent report from Mazandaran province of Iran describes the occurrence of large intracerebral hemorrhage in right hemisphere of an elderly male patient who presented to emergency with loss of consciousness following a 3-day episode of febrile illness [94]. The authors have duly speculated the role of ACE2 receptor activation by the viral particle in disrupting cerebral auto-regulation and thereby resulting in hemorrhage.
The speculation is in agreement with a previous experimental model by Xia et al. which proposed that ADAM-17 mediated brain ACE2 shedding can contribute to neurogenic hypertension [95].
In sum, limited clinical data informs that SARS-CoV-2 has potential to cause CNS vasculopathy, although exact molecular mechanisms are yet to be clarified. Ischemic manifestations, as in other organ systems, are anticipated to be more frequent in CNS as well. Larger studies elucidating CNS vascular manifestations of COVID-19 will be required to have a clearer interpretation of the subject and formulate a therapeutic plan in such circumstances.
SARS-CoV-2 mediated miscellaneous presentation of other systemic vasculopathy
With increasing number of COVID-19 cases around the world, the diversity in presentation among patients is also becoming significant. At the beginning, which was considered to be strictly confined to the lungs is now showing features of multi-organ involvement.
Liver invasion has been reported in a few cases where autopsy revealed micro thrombotic changes in the liver vasculature, along with congestion of the sinuses, hepatocyte degeneration and lobular necrosis with lymphocytic infiltrate [96]. Results like micro-vesicular steatosis, lobular activity in the parenchyma has also been demonstrated on post mortem examination in COVID-19 cases [97].
Laboratory reports have also indicated some degree of liver injury suggested by increased aminotransferase levels in COVID-19 subjects [98]. Sepsis related liver injury which involves hypoxic damage, hepatocellular injury and a distorted immunoregulation may also be attributed to this particular pathogenesis [99]. Ischemic injury to the liver due to thrombosis in the end vessels may cause liver necrosis
Apart from this report stating direct viral invasion of secondary lymphoid organs like spleen and lymph nodes has been noted [100]. Tissue resident CD-169 macrophages expressing the ACE2 receptor seems to be the port of entry in this case. Post mortem examination revealed haemorrhagic hilar lymph nodes with subcapsular sinus histiocytosis.
SARS-CoV-2 may provoke macrophages to release IL-6 up regulating Fas–Fas ligand (Fas-FasL) mediated lymphocytic apoptosis. The picture inside the spleen includes white pulp atrophy, red pulp congestion with depletion of Germinal centres. Taken together this may result in an immunocompromised state which may be an easy access for secondary infections.
Gastrointestinal manifestations are another aspect of the disease drawing attention with growing number of cases. Presence of ACE2 receptors in the gut makes them a potential target for the virus [101]. Diarrhoea, vomiting has precipitated in some of the cases which were confirmed to be SARS-CoV2 infection by stool sample examination [102].
Another case report of a female presenting with abdominal pain and bloody diarrhoea has also been reported [103]. Colonoscopy revealed areas of focal erythema without ulceration in the descending colon and she was diagnosed with haemorrhagic colitis. Investigations were carried out and almost all the possible etiologies of hemorrhagic colitis came back negative.
Given her travel history, her stool samples were sent for SARS-CoV-2 RNA RT polymerase chain reaction which later came back to be positive [103]. With the growing evidence of vascular involvement in COVID-19 patients and a deranged coagulation profile, clinicians should be aware about more fatal complications like mesenteric ischemia [31] or bowel wall necrosis in conjunction with the disease.
Apart from this, the cytokine rage in the disease may lead to a rare hyperinflammatory condition similar to secondary haemophagocytic lymphohistiocytosis (sHLH). HLH is characterised by a febrile cytopenia, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia and elevated cytokine levels. A cytokine profile mimicking that of sHLH in increased IL-2, IL-7, G-CSF, MCP-1, MIP-1α, TNF-α has also been manifested in COVID-19 [22].
Dermatological involvement has also been reported in the form of retiform purpura or live do racemosa consistent with a microvascular occlusive pathology. Histological examination revealed complement meditated injury to the vessel wall [36].
Reference
- Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: origin, transmission, and
- Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423
- Chan JF, To KK, Tse H, Jin DY, Yuen KY (2013) Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol 21:544–555
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S et al (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9:221–236
- Ng WF, To KF, Lam WW, Ng TK, Lee KC (2006) The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1—a review. Hum Pathol 37(4):381–390
- Cheng VC, Hung IF, Tang BS, Chu CM, Wong MM, Chan KH et al (2004) Viral replication in the nasopharynx is associated with diarrhea in patients with severe acute respiratory syndrome. Clin Infect Dis 38:467–475
- Ding Y, Wang H, Shen H, Li Z, Geng J, Han H et al (2003) The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 200:282–289
- Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J et al (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 203:622–630
- Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, Asa SL et al (2005) Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis 191:193–197
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574
- Arabi YM, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer AH et al (2015) Severe neurologic syndrome
- Wang N, Shi X, Jiang L, Zhang S, Wang D, Tong P, Arledge KC (2013) Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res 23(8):986–993
- Abbott CA, Baker E, Sutherland GR, McCaughan GW (1994) Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 40:331–338
- Algahtani H, Subahi A, Shirah B (2016) Neurological complications of middle east respiratory syndrome coronavirus: a report of two cases and review of the literature. Case Rep Neurol Med 2016:3502683
- Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus AD et al (2007) The emerging role of ACE2 in physiology and disease. J Pathol 212:1–11
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J et al (2002) Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277:14838–14843
- Zaim S, Chong JH, Sankaranarayanan V, Harky A (2020) COVID-19 and multi-organ response. Curr Probl Cardiol. https://doi.org/10.1016/j.cpcardiol.2020.100618
- Park WB, Kwon NJ, Choi SJ, Kang CK, Choe PG, Kim JY et al (2020) Virus isolation from the first patient with SARS-CoV-2 in Korea. J Korean Med Sci 35:e84
- Zhang Y, Gao Y, Qiao L, Wang W, Chen D (2020) Inflammatory response cells during acute respiratory distress syndrome in patients with coronavirus disease 2019 (COVID-19). Ann Intern Med. https://doi.org/10.7326/L20-0227
- Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916
- Chen IY, Moriyama M, Chang MF, Ichinohe T (2019) Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol 10:50
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506
- Yang M (2020). Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. Available at SSRN: https://ssrn.com/abstract=3527420 or https://doi.org/10.2139/ssrn.3527420.
- Schett G, Sticherling M, Neurath MF (2020) COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat Rev Immunol 20:271–272
- Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I et al (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34:2
- Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z et al (2020) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin Immunol 214:108393
- Zhang C, Wu Z, Li JW, Zhao H, Wang GQ (2020) The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents 55:105954
- Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK et al (2003) Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326:1358–1362
- Li G, He X, Zhang L, Ran Q, Wang J, Xiong A et al (2020) Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun. https://doi.org/10.1016/j.jaut.2020.102463
- Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao NV, Tzouanas CN et al (2020) SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. https://doi.org/10.1016/j.cell.2020.04.035
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418
- Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M (2018) Endothelial cell functions impaired by interferon in vitro: insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res 163:105–116
- Kaplan MJ, Salmon JE (2011) How does interferon-alpha insult the vasculature? Let me count the ways. Arthritis Rheum 63:334–336
- Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N (2020) Type 1 interferons as a potential treatment against COVID-19. Antiviral Res 178:104791
- Moore JB, June CH (2020) Cytokine release syndrome in severe COVID-19. Science 368:473–474
- Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J et al (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 220:1
- Takahashi K, Ip WE, Michelow IC, Ezekowitz RA (2006) The mannose-binding lectin: a prototypic pattern recognition molecule. Curr Opin Immunol 18:16–23
- Wallis R (2007) Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology 212:289–299
- Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H et al (2020) Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. https://doi.org/10.1101/2020.03.29.20041962
- Liu W, Li H (2020) COVID-19: attacks the 1-beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxi. https://doi.org/10.26434/chemrxiv.11938173.v7
- Hamza I, Dailey HA (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim Biophys Acta 1823:1617–1632
- Zhao J, Yang Y, Huang H, Li D, Gu D, Lu X et al (2020) Relationship between the ABO blood group and the COVID-19 susceptibility. medRxiv. https://doi.org/10.1101/2020.03.11.20031096
- Xiao F, Li X, Wang J, Cao J (2019) Mechanisms of vascular endothelial cell injury in response to intermittent and/or continuous hypoxia exposure and protective effects of anti-inflammatory and anti-oxidant agents. Sleep Breath 23:515–522
- Connors JM, Levy JH (2020) Thromboinflammation and the hypercoagulability of COVID-19. J Thromb Haemost. https://doi.org/10.1111/jth.14849
- Liu F, Xu A, Zhang Y, Xuan W, Yan T, Pan K et al (2020) Patients of COVID-19 may benefit from sustained Lopinavir-combined regimen and the increase of Eosinophil may predict the outcome of COVID-19 progression. Int J Infect Dis 95:183–191
- Leru PM (2019) Eosinophilic disorders: evaluation of current classification and diagnostic criteria, proposal of a practical diagnostic algorithm. Clin Transl Allergy 9:36
- Ramirez GA, Yacoub MR, Ripa M, Mannina D, Cariddi A, Saporiti N et al (2018) Eosinophils from physiology to disease: a comprehensive review. Biomed Res Int 2018:9095275
- Magro CM, Poe JC, Kim C, Shapiro L, Nuovo G, Crow MK et al (2011) Degos disease: a C5b–9/interferon-alpha-mediated endotheliopathy syndrome. Am J Clin Pathol 135:599–610
- Ruffatti A, Calligaro A, Lacognata CS, D’Odorico A, Colpo A, Cardin F et al (2020) Insights into the pathogenesis of catastrophic antiphospholipid syndrome. A case report of relapsing catastrophic antiphospholipid syndrome and review of the literature on ischemic colitis. Clin Rheumatol 39:1347–1355
- Manrique-Caballero CL, Peerapornratana S, Formeck C, Del Rio-Pertuz G, Gomez Danies H, Kellum JA (2020) Typical and atypical hemolytic uremic syndrome in the critically Ill. Crit Care Clin 36:333–356
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S et al (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554–562
- Magro CM, Wang X, Garrett-Bakelman F, Laurence J, Shapiro LS, DeSancho MT (2013) The effects of Eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis 8:185
- Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P et al (2006) A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol 177:4794–4802
- Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR et al (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9:e01753
- Yen YT, Liao F, Hsiao CH, Kao CL, Chen YC, Wu-Hsieh BA (2006) Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol 80:2684–2693
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062
- Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323:1061
- Arachchillage DRJ, Laffan M (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18:1233–1234
- Tang N, Bai H, Chen X, Gong J, Li D, Sun Z (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 18:1094–1099
- Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145
- Bowles L, Platton S, Yartey N, Dave M, Lee K, Hart DP et al (2020) Lupus anticoagulant and abnormal coagulation tests in patients with covid-19. N Engl J Med. https://doi.org/10.1056/NEJMc2013656
- Chen L, Li X, Chen M, Feng Y, Xiong C (2020) The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res 116:1097–1100
- Bilimoria J, Singh H (2019) The Angiopoietin ligands and Tie receptors: potential diagnostic biomarkers of vascular disease. J Recept Signal Transduct Res 39:187–193
- Basu-Ray I, MP. S (2020 ). Cardiac manifestations of coronavirus (COVID-19). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, https://www.ncbi.nlm.nih.gov/books/NBK556152/.
- Hua A, O’Gallagher K, Sado D, Byrne J (2020) Life-threatening cardiac tamponade complicating myo-pericarditis in COVID-19. Eur Heart J 41:2130
- Zeng JH, Liu YX, Yuan J, Wang FX, Wu WB, Li JX et al (2020) First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. https://doi.org/10.1007/s15010-020-01424-5
- Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari R, Shah B et al (2020) ST-segment elevation in patients with covid-19—a case series. N Engl J Med 382:2478
- Fox SE, Akmatbekov A, Harbert JL, Li G, Brown JQ, Vander Heide RS (2020) Pulmonary and cardiac pathology in covid-19: the first autopsy series from New Orleans. medRxiv. https://doi.org/10.1101/2020.04.06.20050575
- (2020). [The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China]. Zhonghua Liu Xing Bing Xue Za Zhi, 41:145-151
- Ruan Q, Yang K, Wang W, Jiang L, Song J (2020) Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan China. Intensive Care Med 46:846
- Han H, Yang L, Liu R, Liu F, Wu KL, Li J et al (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med 58:1116
- Zhang H, Zhou P, Wei Y, Yue H, Wang Y, Hu M et al (2020) Histopathologic changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann Intern Med 172:629
- Xie Y, Wang X, Yang P, Zhang S (2020) COVID-19 complicated by acute pulmonary embolism. Radiology 2(2):e200067
- Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M et al (2020) Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol 33:1007
- Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720
- Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L et al (2020) Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int 97:829–838
- Li Z, Wu M, Yao J, Guo J, Liao X, Song S et al (2020) Caution on kidney dysfunctions of COVID-19 patients. medRxiv. https://doi.org/10.1101/2020.02.08.20021212
- Wang L, Li X, Chen H, Yan S, Li D, Li Y et al (2020) Coronavirus disease 19 infection does not result in acute kidney injury: an analysis of 116 hospitalized patients from Wuhan, China. Am J Nephrol 51:1–6
- Ronco C, Reis T (2020) Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat Rev Nephrol 16:308
- Valizadeh R, Baradaran A, Mirzazadeh A, Baskar VKS (2020) Coronavirus-nephropathy; renal involvement in COVID-19. J Renal Inj Prev 9(2):e18
- Suwanwongse K, Shabarek N (2020) Rhabdomyolysis as a presentation of 2019 novel coronavirus disease. Cureus 12(4):e7561. https://doi.org/10.7759/cureus.7561
- Jin M, Tong Q (2020) Rhabdomyolysis as potential late complication associated with COVID-19. Emerg Infect Dis 26:1618
- Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY et al (2020) Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. https://doi.org/10.1016/j.kint.2020.04.003
- Durvasula R, Wellington T, McNamara E, Watnick S (2020) COVID-19 and kidney failure in the acute care setting: our experience from seattle. Am J Kidney Dis 76:4
- Pan XW, Xu D, Zhang H, Zhou W, Wang LH, Cui XG (2020) Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med 46:1114
- Xu D, Zhang Z, Jin L, Chu F, Mao Y, Wang H et al (2005) Persistent shedding of viable SARS-CoV in urine and stool of SARS patients during the convalescent phase. Eur J Clin Microbiol Infect Dis 24:165–171
- Zhou J, Li C, Zhao G, Chu H, Wang D, Yan HH et al (2017) Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci Adv 3:eaao4966
- Wang R, Liao C, He H, Hu C, Wei Z, Hong Z et al (2020) COVID-19 in hemodialysis patients: a report of 5 cases. Am J Kidney Dis. https://doi.org/10.1053/j.ajkd.2020.03.009
- Mao L, Wang M, Chen S, He Q, Chang J, Hong C et al (2020) Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: a retrospective case series study. medRxiv. https://doi.org/10.1101/2020.02.22.20026500
- Li Y, Wang M, Zhou Y, Chang J, Xian Y, Mao L et al (2020) Acute cerebrovascular disease following COVID- 19: a single centre, retrospective, observational study. Lancet. https://doi.org/10.2139/ssrn.3550025
- Moshayedi P, Ryan TE, Mejia LLP, Nour M, Liebeskind DS (2020) Triage of acute ischemic stroke in confirmed COVID-19: large vessel occlusion associated with coronavirus infection. Front Neurol 11:353
- Zhai P, Ding Y, Li Y (2020) The impact of COVID-19 on ischemic stroke:a case report. Res Square Preprint. https://doi.org/10.21203/rs.3.rs-20393/v1
- Zhou B, She J, Wang Y, Ma X (2020) A case of coronavirus disease 2019 with concomitant acute cerebral infarction and deep vein thrombosis. Front Neurol 11:296
- Sharifi-Razavi A, Karimi N, Rouhani N (2020) COVID-19 and intracerebral haemorrhage: causative or coincidental? New Microbes New Infect 35:100669
- Xia H, Sriramula S, Chhabra KH, Lazartigues E (2013) Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res 113:1087–1096
- Lu Q, Wang R, Qu G, Wang Y, Liu P, Zhu Y et al (2020) General anatomy report of novel coronavirus pneumonia patients. J Forensic Med 36:21–23
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8:420–422
- Li J, Fan JG (2020) Characteristics and Mechanism Of Liver Injury In 2019 Coronavirus Disease. J Clin Transl Hepatol 8:13–17
- Strnad P, Tacke F, Koch A, Trautwein C (2017) Liver—guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol 14:55–66
- Chen Y, Feng Z, Diao B, Wang R, Wang G, Wang C, et al. (2020). The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. medRxiv: 2020.2003.2027.20045427.
- Lattion AL, Soubrier F, Allegrini J, Hubert C, Corvol P, Alhenc-Gelas F (1989) The testicular transcript of the angiotensin I-converting enzyme encodes for the ancestral, non-duplicated form of the enzyme. FEBS Lett 252:99–104
- Yang J, Luo T (2020) First case of covid-19 in the United States. N Engl J Med 382:e53
- Carvalho A, Alqusairi R, Adams A, Paul M, Kothari N, Peters S et al (2020) SARS-CoV-2 gastrointestinal infection causing hemorrhagic colitis: implications for detection and transmission of COVID-19 disease. Am J Gastroenterol. https://doi.org/10.14309/ajg.0000000000000667
More information: Aakriti Gupta et al, Extrapulmonary manifestations of COVID-19, Nature Medicine (2020). DOI: 10.1038/s41591-020-0968-3



{kind=link}