










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Immunotherapy has revolutionized cancer treatment by stimulating the patient’s own immune system to attack cancer cells, yielding remarkably quick and complete remission in some cases.
But such drugs work for less than a quarter of patients because tumors are notoriously adept at evading immune assault.
A new study in mice by researchers at Washington University School of Medicine in St. Louis has shown that the effects of a standard immunotherapy drug can be enhanced by blocking the protein TREM2, resulting in complete elimination of tumors.
The findings, which are published Aug. 11 in the journal Cell, point to a potential new way to unlock the power of immunotherapy for more cancer patients.
“Essentially, we have found a new tool to enhance tumor immunotherapy,” said senior author Marco Colonna, MD, the Robert Rock Belliveau, MD, Professor of Pathology.
“An antibody against TREM2 alone reduces the growth of certain tumors, and when we combine it with an immunotherapy drug, we see total rejection of the tumor. The nice thing is that some anti-TREM2 antibodies are already in clinical trials for another disease.
We have to do more work in animal models to verify these results, but if those work, we’d be able to move into clinical trials fairly easily because there are already a number of antibodies available.”
T cells, a kind of immune cell, have the ability to detect and destroy tumor cells.
To survive, tumors create a suppressive immune environment in and around themselves that keeps T cells subdued.
A type of immunotherapy known as checkpoint inhibition wakes T cells from their quiescence so they can begin attacking the tumor.
But if the tumor environment is still immunosuppressive, checkpoint inhibition alone may not be enough to eliminate the tumor.
An expert on the immune system, Colonna has long studied a protein called TREM2 in the context of Alzheimer’s disease, where it is associated with underperforming immune cells in the brain.
Colonna and first author Martina Molgora, Ph.D., a postdoctoral researcher, realized that the same kind of immune cells, known as macrophages, also were found in tumors, where they produce TREM2 and promote an environment that suppresses the activity of T cells.
“When we looked at where TREM2 is found in the body, we found that it is expressed at high levels inside the tumor and not outside of the tumor,” Colonna said. “So it’s actually an ideal target, because if you engage TREM2, you’ll have little effect on peripheral tissue.”
Colonna and Molgora – along with colleagues Robert D. Schreiber, Ph.D., the Andrew M. and Jane M. Bursky Distinguished Professor; and William Vermi, MD, an immunologist at the University of Brescia – set out to determine whether inhibiting TREM2 could reduce immunosuppression and boost the tumor-killing powers of T cells.
As part of this study, the researchers injected cancerous cells into mice to induce the development of a sarcoma.
The mice were divided into four groups. In one group, the mice received an antibody that blocked TREM2; in another group, a checkpoint inhibitor; in the third group, both; and the fourth group, placebo.
In the mice that received only placebo, the sarcomas grew steadily. In the mice that received the TREM2 antibody or the checkpoint inhibitor alone, the tumors grew more slowly and plateaued or, in a few cases, disappeared. But all of the mice that received both antibodies rejected the tumors completely.
The researchers repeated the experiment using a colorectal cancer cell line with similarly impressive results.
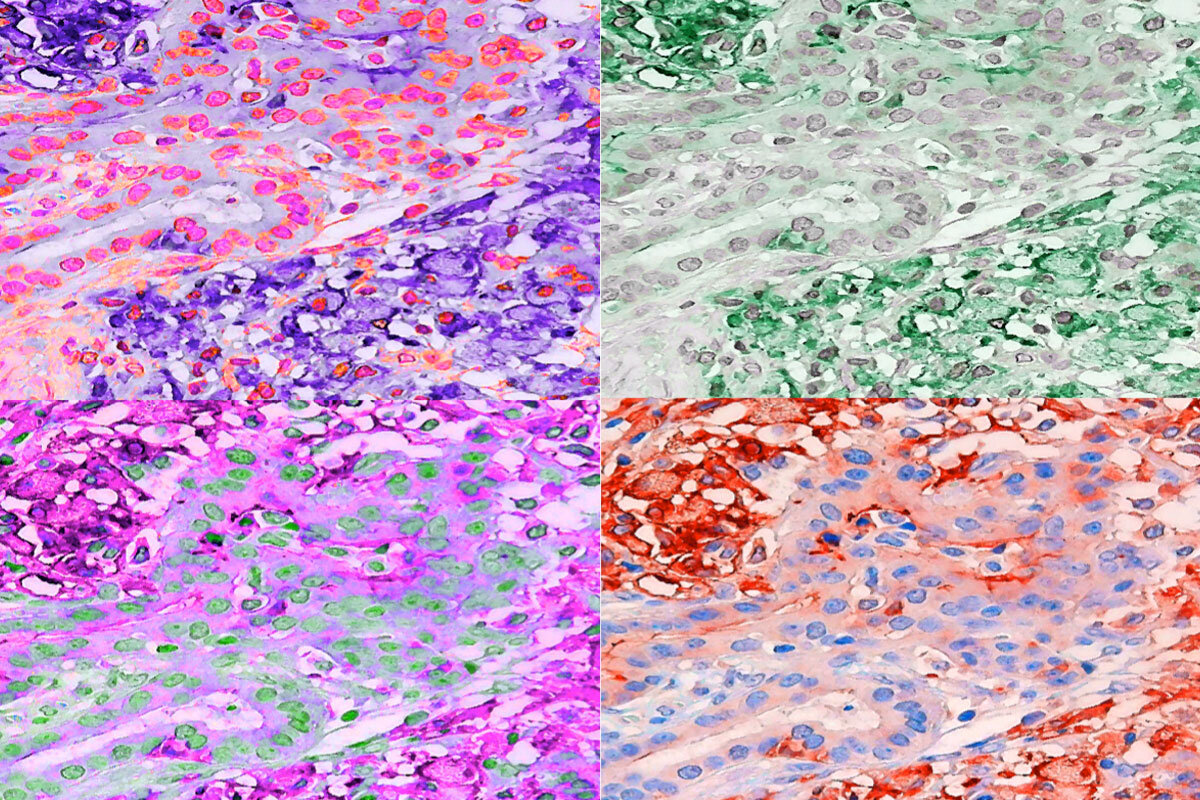
With the help of graduate student Ekaterina Esaulova, who works in the lab of Maxim Artyomov, Ph.D., an associate professor of pathology and immunology, the researchers analyzed immune cells in the tumors of the mice treated with the TREM2 antibody alone.
They found that suppressive macrophages were largely missing and that T cells were plentiful and active, indicating that blocking TREM2 is an effective means of boosting anti-tumor T cell activity.
Further experiments revealed that macrophages with TREM2 are found in many kinds of cancers.
To assess the relationship between TREM2 expression and clinical outcomes, the researchers turned to The Cancer Genome Atlas, a publicly available database of cancer genetics jointly maintained by the National Cancer Institute and the National Human Genome Research Institute.
They found that higher levels of TREM2 correlated with shorter survival in both colorectal cancer and breast cancer.
The researchers are now expanding their study of TREM2 to other kinds of cancers to see whether TREM2 inhibition is a promising strategy for a range of cancers.
“We saw that TREM2 is expressed on over 200 cases of human cancers and different subtypes, but we have only tested models of the colon, sarcoma and breast, so there are other models to test,” Molgora said. “And then we also have a mouse model with a human version of TREM2.”
Added Colonna: “The next step is to do the animal model using the human antibody. And then if that works, we’d be ready, I think, to go into a clinical trial.”
TREMs are type I membrane proteins with an extracellular Ig-like domain and a short cytoplasmic tail that have no intrinsic signaling capacity [1]. TREM signaling relies on association with the DNAX-activating protein 12 (DAP12) cytosolic adapter [2].
DAP12 contains an immunoreceptor tyrosine-based activation motif (ITAM) [1], which becomes phosphorylated upon the activation of DAP12-associated receptors. Phosphorylated ITAM in turn recruits and activates spleen tyrosine kinase (Syk), leading to cellular responses such as the regulation of cytokine production [3,4].
TREM family members play distinct roles in regulating inflammatory responses [1]. For example, the activation of TREM1 amplifies inflammation [5]. In contrast, TREM2 signaling suppresses the inflammatory reaction [1,6,7].
The role of TREM3 in innate immunity has not been elucidated.
TREM2 is a cell surface receptor and a member of the Ig superfamily with one V-type extracellular domain, a charged transmembrane domain, and a short cytoplasmic tail [8].
TREM2 is expressed in monocytes [9], macrophages [10,11], microglia [8,12], dendritic cells (DCs) [13], and osteoclasts [14]. Furthermore, TREM2/DAP12 signaling induces the phosphorylation of extracellular signal-regulated kinase (ERK) and promotes the upregulation of CC chemokine receptor 7 (CCR7) in dendritic cells [13].
In addition, the inhibition of PI3 kinase suppresses TREM2 expression [15]. Although these results suggest that the TREM2/PI3 kinase signaling pathway negatively regulates TREM-2 expression, homeostasis maintenance, TREM2 ligand(s) and TREM2 functions in apoptosis are not clearly understood.
More than 90% of all colon cancers will have an activating mutation of the canonical Wnt signaling pathway, and the dysregulation of the Wnt/β-catenin pathway plays an essential role in colon carcinogenesis [16].
Activated GSK (glycogen synthase kinase) -3β phosphorylates target proteins such as β-catenin, which is a transcription factor that regulates the expression of several genes, including c-myc and cyclin D1, by translocation into the nucleus, where it interacts with transcription factors of the TCF/LEF-1 (T-cell factor/lymphoid enhancer-binding factor family-1) [17,18].
Interestingly, both ERK and Wnt signaling pathways are involved in the regulation of differentiation, organogenesis, adipogenesis, and proliferation, and the aberrant regulation of the Wnt/β-catenin signaling pathway, including the upregulation of β-catenin, is one of the major causes of colon and other cancers [19].
PI3K-activated AKT (protein kinase B) can phosphorylate GSK-3β, leading to the inactivation of GSK-3β and augmentation of β-catenin-TCF4 (T-cell factor 4) transcriptional activity [20].
Recent studies have indicated that the crosstalk between the PI3K/AKT pathway and the GSK-3β/β-catenin pathway and between the ERK pathway and the GSK-3β/β-catenin pathway plays a role in cell survival and proliferation [19,21,22], and TREM2 is one of the molecules that may regulate GSK-3β/β-catenin signaling through these pathways.
However, the underlying mechanisms by which TREM2 regulates Wnt signaling in colon cancer cells are completely unknown.
In this study, we demonstrated that TREM2 inhibits the proliferation of colon cancer cells by attenuating cell cycle progression by downregulating cyclin D1 expression. Furthermore, TREM2 increases β-catenin phosphorylation by activating GSK-3β, which suppresses β-catenin accumulation and TCF4 transcriptional activity, resulting in the downregulation of tumorigenicity in colon cancer cells.
reference link : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6770495/
More information: Cell (2020). DOI: 10.1016/j.cell.2020.07.013



{kind=link}