









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Metformin is the first-line treatment for most cases of type 2 diabetes and one of the most commonly prescribed medications worldwide, with millions of individuals using it to optimise their blood glucose levels.
A new research study, conducted over six years in the Sydney Memory and Ageing Study in 1037 Australians (aged 70 to 90 years old at baseline), has revealed an additional effect: individuals with type 2 diabetes who used metformin experienced slower cognitive decline with lower dementia rates than those who did not use the medication.
The findings provide new hope for a means of reducing the risk of dementia in individuals with type 2 diabetes, and potentially those without diabetes who number nearly 47 million people worldwide.
The study was led by researchers at the Garvan Institute of Medical Research and the Centre for Healthy Brain Ageing (CHeBA), UNSW Sydney, and published in the Journal Diabetes Care.
“We’ve revealed the promising new potential for a safe and widely used medication, which could be life-changing for patients at risk of dementia and their families.
For those with type 2 diabetes, metformin may add something extra to standard glucose lowering in diabetes care: a benefit for cognitive health,” says first author Professor Katherine Samaras, Leader of the Healthy Ageing Research Theme at the Garvan Institute and endocrinologist at St Vincent’s Hospital Sydney.
Protecting brain function
Type 2 diabetes occurs when the body can no longer produce enough insulin to meet its needs, leaving affected individuals unable to maintain blood glucose levels within a normal range.
This can lead to long-term health complications, including cognitive decline.
“As they age, people living with type 2 diabetes have a staggering 60% risk of developing dementia, a devastating condition that impacts thinking, behaviour, the ability to perform everyday tasks and the ability to maintain independence.
This has immense personal, family, economic and societal impacts,” says Professor Samaras.
The researchers of this study investigated data from participants of CHeBA’s Sydney Memory and Ageing Study. In this cohort, 123 study participants had type 2 diabetes, and 67 received metformin to lower blood sugar levels.
The researchers tested cognitive function every two years, using detailed assessments that measured cognition over a number of capabilities, including memory, executive function, attention and speed, and language.
The findings revealed individuals with type 2 diabetes taking metformin had significantly slower cognitive decline and lower dementia risk compared to those not taking metformin.
Remarkably, in those with type 2 diabetes taking metformin, there was no difference in the rate of decline in cognitive function over 6 years compared to those without diabetes.
New use for a common medication
Metformin has been used safely to treat type 2 diabetes for 60 years. It works by reducing the amount of glucose released from the liver into the blood stream and allows the body’s cells to better respond to blood glucose levels.
Studies over the last decade have revealed evidence of metformin’s benefit in cancer, heart disease, polycystic ovary syndrome and weight management.
While the current study suggests metformin may have cognitive benefits for people living with type 2 diabetes, the researchers say it may also benefit those at risk of cognitive decline more broadly.
“This study has provided promising initial evidence that metformin may protect against cognitive decline.
While type 2 diabetes is thought to increase dementia risk by promoting degenerative pathways in the brain and nerves, these pathways also occur in others at risk of dementia and it is possible insulin resistance may be the mediator,” says Professor Samaras.
“To establish a definitive effect, we are now planning a large, randomised controlled trial of metformin in individuals at risk of dementia and assess their cognitive function over three years. This may translate to us being able to repurpose this cheap medication with a robust safety profile to assist in preventing against cognitive decline in older people.”
CHeBA’s Sydney Memory and Ageing Study is an observational study of older Australians that commenced in 2005 and researches the effects of ageing on cognition over time.
Professor Perminder Sachdev, senior author of the study and Co-Director of CHeBA, says: “While an observational study does not provide conclusive ‘proof’ that metformin is protective against dementia, it does encourage us to study this and other anti-diabetic treatments for dementia prevention.
Metformin has even been suggested to be anti-ageing.
The intriguing question is whether metformin is helpful in people in those with normal glucose metabolism. More work is clearly needed.”
BUT…………
Metformin is among the most frequently prescribed drugs worldwide and it is used to facilitate glucose catabolism in patients with impaired insulin signaling (diabetes type 2) (Salani et al., 2014).
Metformin is thought to act by inhibiting mitochondrial respiration (Wheaton et al., 2014). Recently, metformin was tested for additional physiological effects and found to extend life span in animal models ranging from nematodes to mice (Martin-Montalvo et al., 2013; Onken and Driscoll, 2010).
Extensive clinical use in humans enabled the collection and analysis of data on the longevity of human diabetes patients treated with metformin. The metformin-exposed diabetes cohort was found to be longer lived than untreated healthy subjects (Bannister et al., 2014), in line with potential life-prolonging properties of metformin.
Type 2 diabetes is an aging-associated disorder and many patients start metformin treatment in late life. Based on survival analysis of diabetes patients, it was proposed that life prolonging effects of metformin may extend also to metabolic-healthy elderly individuals.
Considering moderate metformin side effects in diabetes and the potential healthy aging benefits, metformin has emerged as an attractive candidate to be clinically tested as the first prospective anti-aging drug in humans.
A short term trial administering metformin to aged (≥65 years old) pre-diabetic humans for a period of 6 weeks had recently been completed and the data was reported (Kulkarni et al., 2018). While effects on pathways such as TOR and immune response were observed, no obvious physiological changes were detected due to short duration of the treatment, leaving long-term effects of metformin on aged healthy humans an open question.
This question is however critical because non-diabetic elderly humans are anticipated to be the first recipients of the putative health span extension treatment with metformin.
Via literature research of animal studies providing evidence of longevity modulation by metformin, we discovered that the pro-survival effect of this drug was mostly studied in young animals or animals exposed to metformin from young adulthood.
The few studies performed in older animals (56-60 week old mice, average lifespan 96 weeks; 8 day old nematodes, average lifespan 14-21 days), either failed to detect life extension by metformin (Alfaras et al., 2017; Anisimov et al., 2011) or revealed toxicity that was partially attributed to the metformin overdose (Cabreiro et al., 2013; Martin-Montalvo et al., 2013; Thangthaeng et al., 2017).
Strikingly, a dose of 50mM metformin which triggered strongest lifespan extension in a seminal study performed in young C. elegans, was moderately toxic when given to middle aged (adulthood day 8) nematodes (Cabreiro et al., 2013; Onken and Driscoll, 2010).
We thus came to a conclusion that the benefits and safety of metformin administration to old non-insulin resistant individuals were not sufficiently investigated, contrary to responses of diabetic patients.
Here we used genetic tests, metabolic measurements, stress reporter assays and omics analyses to detect age-specific effects of metformin in C. elegans and human primary cells. We found that metformin treatment initiated in late life shortens life span and limits cell survival by aggravating aging associated mitochondrial dysfunction towards respiratory failure.
In addition to mitochondrial distortion, old cells failed to enhance the use of glycolysis in response to metformin leading to persistent ATP exhaustion. We found that interventions stabilizing cellular ATP levels, such as ATP repletion and TOR inhibitor rapamycin, alleviate late life metformin toxicity in vitro and in vivo.
We also discovered that early age metformin treatment instigates a range of stress and metabolic adaptations which likely underlay longevity extension by metformin. Importantly, the induction of these favorable responses was strongly impaired in late life.
Particularly, we show that early but not late life metformin treatment induces a lipid turnover response similar to dietary restriction (DR). We also found that this early life DR mimetic phenotype is instigated by the triglyceride lipolysis pathway regulated by the protein kinase A and not by AMP activated protein kinase (AMPK) pathway, as suggested previously.
Subsequently, we showed that metformin treatment restricted to early adulthood is sufficient for life extension, strengthening the key role of early life stress and metabolic adaptations in longevity benefits of metformin. Finally, we demonstrate that daf-2(e1370) mutants, carrying diabetes-like insufficiency of the C. elegans insulin receptor, are resilient to late life metformin toxicity in comparison to age-matched wild type controls, due to improved capacity to sustain ATP synthesis during old age metformin exposure. Collectively, we uncovered an alarming capability of metformin to induce metabolic failure in non-insulin resistant old subjects which may limit its benefits for non-diabetic elderly humans.
Results
Late life metformin treatment is detrimental for longevity
To address the outcomes of metformin treatment at different age, we treated young adult (3 days old, day 1 of adulthood), adult at the age of reproduction decline (day 4 of adulthood), middle aged (day 8 of adulthood) and old (day 10 of adulthood) wild type C. elegans worms with different doses of metformin – 10mM, 25mM and 50mM. 50mM metformin is the common dose used to induce lifespan extension in C. elegans while 10mM is the lowest dose linked to reproducible life extension in this model in previous reports (Cabreiro et al., 2013; Onken and Driscoll, 2010; Pryor et al., 2019).
We found that metformin treatment started at young age (days 1 and 4 of adulthood) extended lifespan of nematodes at all doses used (Figure 1A and Figure S1A).
Within treatment initiated on day 8 of adulthood, the doses of 50mM and 25mM metformin reduced median lifespan but extended maximal lifespan consistent with previous observations (Cabreiro et al., 2013) while 10mM dose was longevity-extending with no detrimental effects (Figure S1B).
Strikingly, on day 10 of adulthood metformin was toxic at all doses used with a large proportion of drug-exposed animals dying within first 24 hours of treatment (Figure 1B). Our first experiments in nematodes thus revealed an evident age-dependent decrease in metformin tolerance which culminated in late life toxicity of all metformin doses tested, indicating possible safety risks of late life metformin administration.

Metformin treatment initiated in late life exerts toxicity and limits survival independently of AMPK and microbial changes.
Wild type (WT, N2 Bristol strain) nematodes were treated with indicated doses of metformin (Met) on day 1 (A) and day 10 (B) of adulthood (AD1 and AD10 respectively), survival was scored daily. WT nematodes were grown on alive and UV-killed HT115 (C) and OP50 (D) E. coli and treated with 50mM metformin on AD10. Survival was scored daily. WT (E, left) and AMPK deficient (E, right) nematodes were treated with 50mM metformin on AD1 and AD10, survival was scored daily. Significance was measured by log-rank test, n≥100 in all cases, the exact n numbers and statistical values for all panels are presented in Table S1. Each experiment was repeated ≥3 times; one representative result is shown in all cases.
Late life metformin toxicity is independent of microbiome
To understand the mechanism of late life metformin toxicity, we first addressed known pathways regulating lifespan extension by this compound. The pro-longevity effect of metformin in young C. elegans nematodes was previously linked to changes of microbial metabolism induced by this drug (Cabreiro et al., 2013).
To test if old age metformin toxicity relied on similar microbiome alterations we treated worms with metformin in the presence of living and UV-killed OP50 (metformin sensitive) and HT115 (metformin-resistant) E. coli strains.
Old age metformin toxicity developed regardless of the bacterial viability and/or strain (Figure 1C and D) suggesting that late life metformin intolerance is independent of previously uncovered microbiome changes. Interestingly, the baseline survival of nematodes differed between UV killed OP50 and HT115 diets in line with recently reported dependence of nematode physiological behaviors on the bacterial source (Revtovich et al., 2019).
AMPK is not required for late life toxicity of metformin
Another component essential for young age life-extending effect of metformin is AMP-activated protein kinase (AMPK); particularly metformin failed to promote longevity in nematodes lacking AMPK orthologue AAK-2 (Onken and Driscoll, 2010). In order to probe the requirement of AMPK for old age toxicity of metformin we treated young and old wild type and aak-2(ok524) mutant animals with this drug. Consistent with previous reports, early life metformin treatment was unable to induce lifespan extension in aak-2 deficient worms (Figure 1E); at the same time late life metformin toxicity did develop in mutant animals, indicating that life shortening induced by metformin at old age is not executed by AMPK.
Metformin toxicity is triggered by mitochondrial impairments
One of metformin’s primary functions is to inhibit complex I of the mitochondrial electron transport chain (ETC), affecting mitochondrial membrane potential and ATP production (Andrzejewski et al., 2014; Cameron et al., 2018; Wheaton et al., 2014).
Growth inhibition by metformin was previously linked to impaired mitochondrial respiration in nematodes and mammalian cells (Wu et al., 2016). Additionally, the accumulation of damaged and dysfunctional mitochondria, which may enhance the negative impact of ETC complex I inhibitors on cell survival, is one of the best characterized hallmarks of aging (Bratic and Larsson, 2013; Bratic and Trifunovic, 2010; Cellerino and Ori, 2017; Sun et al., 2016; Taylor and Dillin, 2011).
Mitochondrial deterioration comparable to aging occurs prematurely in mutant animals, defective in mitochondrial biogenesis and quality control (Sun et al., 2016; Trifunovic et al., 2004). To test if metformin toxicity during aging (and metformin toxicity in general) is driven by accumulation of mitochondrial impairments, we obtained mutants harboring deficiencies of diverse mitochondrial homeostasis pathways: mitochondrial unfolded protein response (atfs-1(gk3094)) (Nargund et al., 2012), mitochondrial biogenesis (skn-1(zj15)) (Palikaras et al., 2015), mitochondrial respiration (isp-1(qm150)) (Feng et al., 2001) and mitochondrial protein quality control (ubl-5(ok3389)) (Benedetti et al., 2006), and treated these animals with metformin along with wild type counterparts.
A combination of metformin with congenital mitochondrial impairments led to an early life onset of metformin toxicity in all mutant backgrounds tested (including normally long-lived isp-1(qm150) mutants) clearly linking metformin intolerance to elevated abundance of dysfunctional mitochondria (Figure 2A-C, Figure S2A).
The same effect (premature onset of metformin toxicity at young age) was observed in nematodes and human primary cells incubated with mitochondrial uncoupling agent carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) along with metformin administration (Figure 2D-E and Figure S2B).
Of note, congenital mitochondrial respiration defects were recently found to limit the life extending interplay between metformin and the microbiome (Pryor et al., 2019) in line with the key role of mitochondrial integrity in diverse longevity benefits of metformin.
To test if mitophagy and/or mitochondrial unfolded protein response (UPR MT) – the protective pathways responding to mitochondrial failure and known to deteriorate during aging (Sun et al., 2016), were directly induced by metformin we measured the abundance of mitochondrial proteins and the expression of hsp-6::gfp transgene (UPR MT reporter) in young and old animals treated with this drug.
At both ages metformin administration didn’t lead to either elevated expression of GFP or reduction of mitochondrial protein levels (Figure S3A-C), indicating that mitophagy and UPR MT are not prominently triggered by metformin and likely play no role in immediate cellular adaptation to metformin-induced effects.
Of note, lack of UPR MT induction by metformin has previously been reported by an independent group (De Haes et al., 2014). We thus show that the early onset of metformin toxicity in mitochondrial mutants is likely driven by the accumulation of mitochondrial damages prior to metformin administration, comparable to what occurs during aging.

Metformin toxicity associates with ATP exhaustion and is alleviated by ATP repletion
To measure direct age-specific effects of metformin on mitochondrial performance and to test the conservation of our findings in humans, we analyzed the impact of metformin treatment on homeostasis of early passage (young) and late passage (old, replicative senescent) primary human skin fibroblasts.
The replicative senescence model was chosen because of its high relevance for normal human aging as senescent cells accumulate in aging tissues. In vitro aging was performed according to standard procedures demonstrated to yield cells carrying key hallmarks of aging (Tigges et al., 2014), and aging-associated mitochondrial decline of late passage cells was verified by the Seahorse analysis (Figure 3C-D, Figure S4A and C).
We found that, at high doses, metformin was toxic to both young and old cells but old cells (similar to old nematodes) showed a much stronger viability decline and succumbed to toxicity already at lower doses of metformin (Figure 3A-B).
The oxygen consumption rate (OCR) measurements demonstrated a significant effect of metformin on basal respiration in young and old cells (Figure 3C, Figure S4A), while inhibition of mitochondrial ATP synthesis by metformin was stronger in old fibroblasts (Figure S4C).
In addition, only old cells showed a decrease of maximal respiration in response to metformin (Figure 3D, Figure S4A) consistent with a stronger negative impact of this drug on mitochondria of old cells. Interestingly, the extracellular acidification rate (ECAR) was potently increased in young metformin treated fibroblasts (Figure 3E, Figure S4B) in line with their elevated reliance on glycolysis in response to mitochondrial insufficiency triggered by metformin.
This adaptive increase of glycolysis was markedly reduced in old metformin exposed cells (Figure 3E, Figure S4B), depriving these cells, in combination with the stronger mitochondrial hindrance by metformin, of effective pathways of ATP synthesis. Subsequent determination of cellular ATP content indeed showed a stronger decline of ATP levels in old metformin treated cells (Figure 3F).
We also detected a stronger distortion of the mitochondrial membrane potential in old compared to young metformin exposed fibroblasts (Figure 3G), in line with a more potent mitochondrial decline observed in old cells via oxygen consumption analysis.

We next measured the effect of metformin on organismal ATP content in nematodes. Metformin treatment of young animals didn’t cause negative changes of systemic ATP levels (Figure 3H), consistent with reduced mitochondrial impairments and intact metabolic adaptability at young age.
Strikingly, old animals showed a strong reduction of baseline ATP levels, compared to untreated young animals, followed by a further 76% decline of ATP content induced by metformin (Figure 3H, Table S2). These results are consistent with known mitochondrial deterioration and reduced energetics of old animals (and cells) (Brys et al., 2010; Drew et al., 2003), and suggest, along with above reported age-specific metabolic phenotypes, that metformin toxicity may be linked to a failure of old cells to maintain ATP synthesis during metformin treatment, leading to a decline of ATP content down to levels incompatible with cell viability.
To test this hypothesis we asked if ectopic ATP supplementation would rescue metformin toxicity. Providing ATP to animals is not feasible due to insufficient bioavailability upon oral supplementation (Arts et al., 2012), we were however able to supplement ATP to fibroblasts in culture using millimolar concentrations as previously described (1978).
Ectopic ATP repletion alleviated cell death, ATP exhaustion and loss of mitochondrial membrane potential induced by metformin (Figure 3I-J, Figure S5A-D) and by another complex I inhibitor rotenone (Figure S6A-C) linking viability loss upon treatment with ETC complex I inhibitors to alterations in energy homeostasis.
The rescue of the mitochondrial membrane potential by ATP repletion was consistent with the previously reported ability of the mitochondrial ATP synthase to restore membrane potential under conditions of respiratory failure by consuming ATP (Chinopoulos and Adam-Vizi, 2010).
Metformin toxicity is suspended in nematodes carrying insulin receptor insufficiency
Because metformin intolerance, comparable to our findings, has not been reported in type 2 diabetes patients receiving treatment in late life, we decided to test the old age metformin response of nematodes lacking functional insulin receptor (a molecular mimetic of insulin resistance in type 2 diabetes).
For that we chose a well characterized daf-2(e1370) strain which carries a temperature sensitive mutation of the C. elegans IGF-1/Insulin receptor orthologue DAF-2 (Dorman et al., 1995). At a restrictive temperature of 20°C daf-2(e1370) mutants exhibit partial loss of insulin receptor function leading to a variety of phenotypes, partially comparable to diabetic patients (such as enhanced fat deposition) (Kimura et al., 1997; Tissenbaum and Ruvkun, 1998).
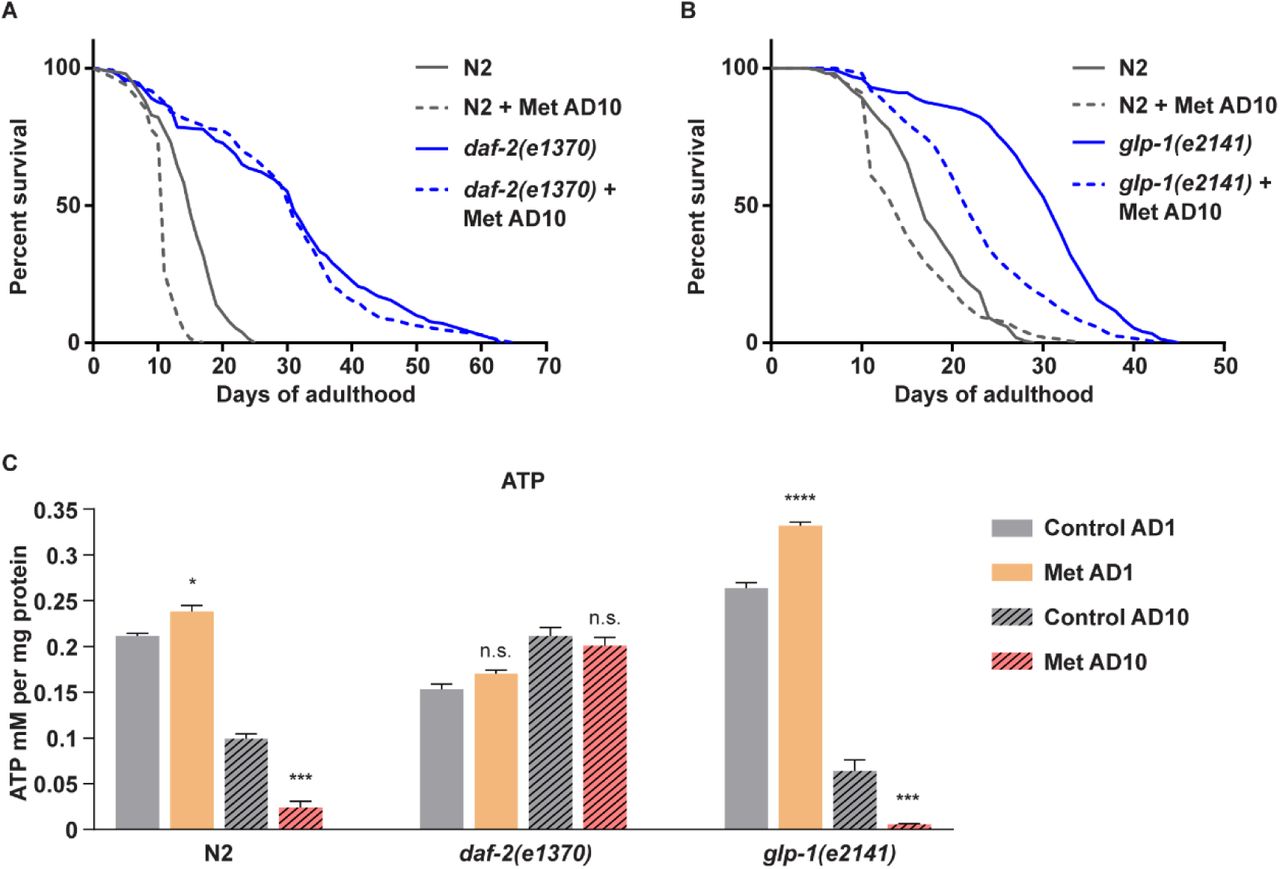
We next measured survival of daf-2(e1370) and wild type nematodes treated with metformin on day 10 of adulthood (AD10) and observed a striking resilience of the mutants to metformin killing at this age (Figure 4A). Systemic ATP measurements revealed a moderate reduction of baseline ATP synthesis in young daf-2(e1370) mutants (Figure 4C) consistent with previous reports (Brys et al., 2010; Palikaras et al., 2015).
Interestingly neither aging nor metformin had a negative impact on ATP levels in AD10 daf-2(e1370) animals, contrary to the response of wild type controls (Figure 4C), associating the resilience of the mutants to metformin toxicity with their ability to sustain ATP synthesis during late life metformin treatment. Of note, the enhanced competence of daf-2(e1370) mutants in ATP synthesis during late life is also consistent with previous reports (Brys et al., 2010; Palikaras et al., 2015).
Importantly, the resistance of daf-2(e1370) animals to metformin wasn’t infinite and mutant nematodes exposed to the drug on adulthood day 21 (AD21) showed levels of metformin toxicity comparable to AD10 wild type controls (Figure S7A), indicating that the insulin receptor itself is not required for the induction of late life metformin toxicity.
In summary, these data reiterate the key role of ATP exhaustion in late life metformin toxicity and indicate that inhibition of insulin receptor signaling, comparable to insulin resistance in diabetes, provides extended protection from late life metformin intolerance.
Metformin resilience of insulin receptor mutants is not due to their slower rate of aging
The daf-2(e1370) mutants are known to be long-lived in comparison to wild type animals (Dorman et al., 1995; Kimura et al., 1997; Tissenbaum and Ruvkun, 1998). To ensure that the prolonged metformin resilience of daf-2(e1370) animals is not a simple consequence of their slower rate of aging, we selected a different comparably long-lived strain – germline-deprived glp-1(e2141) mutants (Lapierre et al., 2011), and treated these animals with metformin on day 10 of adulthood (AD10).
Unlike daf-2(e1370) nematodes, glp-1(e2141) worms succumbed to AD10 metformin killing similar to wild type controls (Figure 4B). They also showed both aging-triggered and metformin-aggravated declines of ATP levels, unlike AD10 daf-2(e1370) mutants and similar to wild type controls (Figure 4C).
We thus determined that the ability to sustain ATP synthesis in late life and the corresponding metformin resilience are not common phenotypes of all long-lived mutants but rather a specific feature of insulin receptor deficient animals.
Metformin resilience of insulin receptor mutants depends on sustained ATP synthesis in late life regulated by DAF-16/FOXO
The exact mechanism behind the enhanced metabolic competence of daf-2(e1370) mutants in late life remains to be elucidated. However, similar to most of daf-2(e1370) beneficial phenotypes, this capacity may depend on the FOXO transcription factor DAF-16 (Kenyon, 2011; Palikaras et al., 2015). Consistently, daf-2(e1370); daf-16(mu86) double mutants didn’t show metformin resilience on adulthood day 10, unlike daf-2(e1370) single mutants and similar to wild type controls (Figure S7B).
The double mutants also showed a clear decline of ATP synthesis upon AD10 metformin treatment (Figure S7C) in line with the requirement of DAF-16 for metabolic resilience of the daf-2(e1370) genetic background. Because DAF-16 is an important mediator of multiple stress adaptations (Kenyon, 2011), we tested if the response to metformin required DAF-16 directly by measuring nuclear translocation of the DAF-16::GFP fusion protein during metformin or heat shock treatment (used as positive control).
While DAF-16 was clearly translocating into the nucleus in response to heat stress, little translocation (above negative control values) could be observed in the course of metformin treatment regardless of age (Figure S8A-C), suggesting that metformin resilience of daf-2(e1370) mutants is not driven by the direct involvement of DAF-16 in stress adaptations during metformin treatment.
The capability of daf-2(e1370) animals to sustain stable ATP synthesis at old age has previously been linked to elevated mitochondrial turnover induced by life-long subtitle metabolic stress, leading to improved mitochondrial quality in late life (Brys et al., 2010; Palikaras et al., 2015). Our proteomics measurements indeed revealed reduced baseline levels of mitochondrial proteins in daf-2(e1370) mutants compared to wild type animals (Figure S7D) consistent with elevated mitochondrial turnover.
Interestingly, mitochondrial protein levels of daf-2(e1370); daf-16(mu86) double mutants were similar to values observed in wild type animals (Figure S7D), suggesting that mitochondrial replenishing triggered by insulin receptor deficiency is inhibited in the absence of DAF-16.
Consistent with the key role of DAF-16 in supporting metabolic homeostasis of daf-2(e1370) mutants, the daf-2(e1370); daf-16(mu86) double mutants demonstrated strong impairments of baseline ATP synthesis already at young age (Figure S7C).
Metformin rapidly activates diverse longevity assurance pathways in young but not old animals
To test if other metformin responses, in addition to ATP synthesis, become altered in late life we performed an unbiased proteomics analysis of animals treated with metformin on days 1 and 10 of adulthood for 24 and 48 hours, and compared these to age- and time point-matched untreated controls.
The abundance of only a restricted proportion of protein groups was affected by metformin at both ages and at both times of treatment (seen by comparing numbers of altered proteins (circled) and non-altered proteins (outer box) in Figures 5A and S9A), highlighting the specificity of the response elicited by the drug treatment.
By comparing the protein expression profiles obtained (4572 protein groups were quantified in total, Table S3), we determined that global responses to metformin at young and old age were largely distinct, as indicated by a limited overlap between commonly affected proteins at both 24 and 48 hours of treatment (Figure 5A and S9A), and a modest correlation between induced protein changes (Figure 5B and S9B).
Interestingly, the similarity between young and old responses was stronger at 48 hours of treatment, which could be explained either by a delay of old animals in responding to the drug or by an enrichment of the 48h old age metformin-exposed sample with animals that were most competent in their adaptation to metformin treatment (based on our survival analyses a significant proportion of metformin sensitive old animals died between 24 and 48h of treatment).

By analyzing pathways regulated by metformin at distinct age points, we detected a rapid activation of adaptive stress responses and longevity assurance pathways such as downregulation of the ribosome (MacInnes, 2016) and induction of oxidative stress response (Honda and Honda, 1999), immune response (Xia et al., 2019) and heat stress response (Baldi et al., 2017) in young animals (Figure 5C-E and S9C). General autophagy (Hansen et al., 2018) was also comparably regulated albeit to a smaller extent (Figure S9D).
These data are consistent with previous studies suggesting that metformin extends life span by inducing stress adaptations such as oxidative stress response (De Haes et al., 2014; Onken and Driscoll, 2010).
Unlike previous reports, however, our data demonstrates for the first time that metformin simultaneously triggers a complex array of diverse stress responses which likely all contribute to life extension by this drug.
Strikingly, the induction of stress adaptations by metformin was either inhibited or delayed in old animals (Figure 5C-E, Figure S9C-D) suggesting that, in addition to metformin-triggered ATP exhaustion, also the longevity assurance component of the metformin response is impaired in late life.
To investigate the age specificity of metformin-triggered stress responses by an independent method, we measured the induction of general autophagy by metformin in a transgenic strain expressing the LGG-1::mCherry fusion protein (Gosai et al., 2010).
LGG-1 is a C. elegans LC3 orthologue that gets recruited to autophagosome membranes during the autophagy process, giving rise (in a reporter setting) to distinct puncta which can be quantified microscopically (Figure S10A).
Autophagy was chosen for the validation test because of its known antagonistic effect on longevity depending on age (Wilhelm et al., 2017) and mitochondrial integrity (Zhou et al., 2019).
By treating young and old transgenic animals with metformin, we could indeed observe a substantial induction of autophagosome formation in young but not old metformin-exposed animals (Figure S10B-C) with effects size comparable to the proteomics data (Figure S9D).
Interestingly, the absolute numbers of autophagy puncta in control untreated animals were significantly higher at old age (Figure S10D-E) consistent with the previously observed inhibition of autophagy flux during late life (Wilhelm et al., 2017).
The specificity of observed puncta to the autophagy process was confirmed by exposing transgenic animals to RNAi against key autophagy mediator beclin 1 (bec-1) (Figure S10F).
In summary, we uncovered that metformin treatment exerts an array of stress adaptation responses in young animals while old animals fail to activate these signals to a comparable extent in line with previous hints of impaired stress resilience at old age (Haigis and Yankner, 2010).
To probe the contribution of early life adaptive events to the pro-longevity effect of metformin we exposed nematodes to the drug during early adulthood and indeed found that metformin treatment restricted to young age is sufficient to confer in vivo life span benefits (Figure S10G).
Old age specific metformin response is enriched in distinct mediators of lipid metabolism
We next screened the proteomics data for pathways activated by metformin predominantly in old animals. We found that peroxisomal components along with mitochondrial and peroxisomal enzymes implicated in fatty acid β-oxidation (such as acs-1, acs-2, acox-2) (Zhang et al., 2011) were upregulated in old stronger than in young animals (Figure 5F, H).
Also proteins associated with lipid droplets, serving as key lipid storage units in C. elegans, (vitellogenins, dehydrogenases) (Vrablik et al., 2015; Zhang et al., 2012) were upregulated in old while they were downregulated in young metformin treated animals (Figure 5G, Figure S9E).
In addition, ribosomal components found to be part of the lipid droplet proteome (Vrablik et al., 2015; Zhang et al., 2012) were downregulated in young but not in old metformin exposed nematodes (Figure S9C). Collectively, our data indicated that metformin treatment leads to an increase in peroxisomal content, which is stronger at old compared to young age, while lipid droplet components are downregulated in young and upregulated in old animals.
Interestingly, the downregulation of lipid droplets and the upregulation of peroxisomes by metformin in young animals were recently reported by an independent study (Pryor et al., 2019) which however didn’t address the age specificity of these phenotypes.
Metformin treatment leads to distinct lipid turnover responses in young and old animals
Because proteomics analysis revealed the age-specific regulation of lipid turnover mediators by metformin, we asked if metformin affects systemic lipid deposition in an age-dependent manner.
By performing Oil Red O whole body lipid staining in C. elegans we detected a significant decline of lipid levels in young animals exposed to metformin while no such decline could be observed in old animals (Figure 6A, Figure S12A).
Importantly, a reduction of whole body lipid content by metformin is consistent with its role as a dietary restriction (DR) mimetic (Onken and Driscoll, 2010) which was recently found to be essential for the longevity extension by this drug (Pryor et al., 2019). Our data in old animals demonstrates that this beneficial DR effect of metformin is abolished during aging.

We next performed lipidomics analysis by mass spectrometry in young and old nematodes exposed to metformin to characterize the age-specific lipid turnover phenotype induced by this drug in a greater detail. By comparing lipid abundancies in untreated young and old worms we observed a significant exhaustion of phospholipids (PLs), free fatty acids (FFAs), lysophospholipids and polyunsaturated fatty acid (PUFA)-rich lysophospholipids which occurred due to aging (Figure S13A-B and Table S4).
Of note, these observations are consistent with a recent lipidomics report characterizing basal lipid changes in aging C. elegans (Gao et al., 2017). Interestingly, the levels of triglycerides (TAGs) were markedly increased in old animals compared to young worms (Figure 6B, D and Table S4), in contrast to other lipid classes measured and reminiscent of the aberrant accumulation of triglycerides observed in human aging (Cree et al., 2004) and upon mitochondrial insufficiency (Vankoningsloo et al., 2006). Importantly, the accumulation of TAGs during aging correlated with the elevated whole body lipid content detected by Oil Red O staining in old untreated nematodes (Figure 6A).
We next compared lipid responses to metformin in young and old animals and found that in young nematodes metformin lowered the abundance of diverse lipid subclasses including free fatty acids and triglycerides, consistent with the DR-like phenotype (Figure 6B-D, Figure S13C-E, Table S4).
Strikingly, the levels of triglycerides, including TAGs containing highly unsaturated PUFAs, were further elevated in old metformin exposed animals (Figure 6B, D and Figure S13E), contrary to the response of young nematodes and consistent with the loss of the metformin DR effect at old age.
The opposite changes in triglyceride content in young and old animals exposed to metformin were also consistent with the opposite dynamics of lipid droplet-associated proteins observed in these animals by proteomics (Figure 5G, Figure S9C, E): triglycerides are the key components of lipid droplets, and lipid droplet turnover supports the engagement of TAGs during fasting and DR (Lee et al., 2014).
Along with the increase in triglycerides and highly unsaturated PUFA-rich triglycerides (Figure S13E), metformin triggered a reduction of long-chain and PUFA-rich lysophospholipids and free fatty acids in old animals (Figure S14A-C and Table S4), similar to aberrant lipid rearrangements observed during persistent metabolic stress (Markel et al., 1985; Nguyen et al., 2017; Steinhauser et al., 2018) and in line with the differential abundance of distinct lipid turnover mediators detected in metformin-exposed old nematodes by proteomics.
In summary, old animals failed to develop a DR mimetic lipid turnover phenotype in response to metformin, contrary to young worms, and rather showed an exacerbation of pre-existing aging-associated lipid abnormalities upon metformin treatment.
Given the recently reported key importance of lipid turnover, resembling DR, for life extension by metformin (Pryor et al., 2019), the reversal of this DR response during aging likely contributes to the lack of metformin benefits in late life.
Metformin-triggered lipid changes are driven by distinct mechanisms at young and old age
Previous studies implicated AMPK in lipid changes linked to aging and mitochondrial alterations (Gao et al., 2017; Weir et al., 2017).
AMPK has also been linked to a reduction of de-novo lipid synthesis in metformin-exposed hepatocytes, muscle and liver (Boudaba et al., 2018; Collier et al., 2006; Fullerton et al., 2013; Zang et al., 2004). We next asked if lipid transformations triggered by metformin at young or old age were mediated by AMPK.
Wild type and AMPK (AAK-2) deficient animals were treated with metformin at young and old age followed by Oil Red O whole body lipid staining. We found that old AMPK deficient animals accumulated additional lipids in response to metformin while no such changes were seen in wild type nematodes (Figure S11A, S12C), in line with an important role of AMPK in mitigating the aberrant lipid accumulation during late life metabolic stress.
But curiously, young AMPK deficient nematodes demonstrated an even stronger lipid reduction in response to metformin compared to wild type controls (Figure 6E, S11C and S12B) indicating that AMPK is not the primary instigator of early life metformin DR response but rather plays an inhibitory role in this process.
Because the age-specific lipid turnover induced by metformin affected triglycerides stronger than other lipid species, we next asked if the triglyceride lipolysis pathway regulated by protein kinase A (PKA) in response to fasting (Lee et al., 2014) is the principle driver of early life metformin effect on lipid reserves.
By performing Oil Red O whole body lipid staining in control nematodes and in nematodes harboring RNAi knock down of kin-1 (the C. elegans orthologue of PKA) or atgl-1 (the orthologue of the adipose triglyceride lipase, the key effector of the PKA lipolysis pathway) we found that inactivation of the PKA pathway indeed suppressed the DR-like lipid turnover induced by metformin in early life (Figure 6F, S11B and S12D-E).
Interestingly, AMPK was previously shown to directly modulate the activity of ATGL-1 towards a more moderate turnover of triglyceride reserves, and this inhibitory capacity of AMPK was found to be essential for the extended longevity of long-lived C. elegans dauer larvae (Narbonne and Roy, 2009).
In our tests we observed a stronger decline of lipid content in young AMPK deficient animals exposed to metformin compared to wild type controls (Figure S11C), it is thus feasible that the modulatory effect of AMPK on the PKA lipolysis pathway is behind the known essential role of AMPK in longevity extension by metformin (Onken and Driscoll, 2010), by preventing the untimely lipid exhaustion during early life metformin exposure.
Importantly, young animals exposed to metformin in the presence of HT115 E. coli (the RNAi vehicle), a condition previously found to be deprived of metformin life extension similar to AMPK mutants (Cabreiro et al., 2013), showed markedly enhanced loss of lipids at 24h of drug treatment compared to OP50 E. coli control and similar to AMPK-deficient animals (Figure S11D and S12D-E, empty vector control); this excessive loss of lipids was clearly prevented by the inactivation of ATGL-1, the key triglyceride lipase regulated by AMPK (Kim et al., 2016)(Figure S11D and S12D).
Our data thus supports the model of metformin life extension where prevention of untimely and unhealthy lipid loss is as important for improved longevity as the induction of the beneficial DR response, previously linked to metformin induced life prolongation (Onken and Driscoll, 2010; Pryor et al., 2019).
We also show that AMPK has age-specific roles in metformin-triggered lipid turnover and that the PKA and not AMPK pathway instigates the DR-like lipid utilization response in young metformin treated animals.
ATP exhaustion and late life metformin toxicity are alleviated by in vivo rapamycin co-treatment
Because appropriate lipid turnover is essential for supporting the mitochondria with metabolites required for effective ATP synthesis, our molecular analysis of key metformin-modulated pathways further supported the primary role of ATP exhaustion in cellular deterioration triggered by metformin in late life.
We next decided to test if accessible in vivo interventions which stabilize cellular ATP levels alleviate late life metformin toxicity. One known intervention that stabilizes ATP content under conditions of mitochondrial dysfunction and carbon starvation is TOR inhibitor rapamycin (Thomsson et al., 2005; Zheng et al., 2016).
Interestingly TOR inhibition was previously found to be essential to support nematode development and cell survival upon treatment with toxic high doses of metformin (Wu et al., 2016). Previous studies also indicated that rapamycin co-exposure enhances longevity benefits of early life metformin treatment (Strong et al., 2016).
We next exposed pre-senescent fibroblasts to metformin in the presence or absence of rapamycin and observed a clear alleviation of metformin toxicity in rapamycin pre-treated cells (Figure 7A and S15A-B) accompanied by blunted ATP exhaustion and reduced loss of mitochondrial membrane potential in these cells (Figure 7B-C and S15C-D).
To test if rapamycin co-exposure protects against late life metformin toxicity also in vivo, we pre-treated nematodes with rapamycin from adulthood day 8 (AD8) followed by metformin treatment on adulthood day 10. Consistent with our cell culture observations, rapamycin co-treatment significantly alleviated late life metformin intolerance as seen by improvement of both median and maximal life spans in rapamycin/metformin co-exposed animals (Figure 7D).
These in vivo findings confirm the decisive role of ATP exhaustion in late life metformin toxicity and suggest that interventions capable of stabilizing cellular ATP content, such as rapamycin administration, could be utilized for prevention of metformin intolerance during aging. The incomplete reversal of late life metformin toxicity by rapamycin is in line with the limited capacity of this drug to preserve cellular ATP levels.

reference link: https://www.biorxiv.org/content/10.1101/863357v1.full



{kind=link}