










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



La metformina è il trattamento di prima linea per la maggior parte dei casi di diabete di tipo 2 e uno dei farmaci più comunemente prescritti in tutto il mondo, con milioni di persone che la utilizzano per ottimizzare i livelli di glucosio nel sangue.
Un nuovo studio di ricerca, condotto in sei anni nel Sydney Memory and Aging Study in 1037 australiani (di età compresa tra 70 e 90 anni al basale), ha rivelato un effetto aggiuntivo: gli individui con diabete di tipo 2 che hanno usato metformina hanno sperimentato un declino cognitivo più lento con tassi di demenza rispetto a coloro che non hanno usato il farmaco.
I risultati forniscono una nuova speranza per un mezzo per ridurre il rischio di demenza negli individui con diabete di tipo 2 e potenzialmente in quelli senza diabete che contano quasi 47 milioni di persone in tutto il mondo.
Lo studio è stato condotto dai ricercatori del Garvan Institute of Medical Research e del Centre for Healthy Brain Ageing (CHeBA), UNSW Sydney, e pubblicato sul Journal Diabetes Care.
“Abbiamo rivelato il nuovo e promettente potenziale di un farmaco sicuro e ampiamente utilizzato, che potrebbe cambiare la vita dei pazienti a rischio di demenza e delle loro famiglie.
Per le persone con diabete di tipo 2, la metformina può aggiungere qualcosa in più alla riduzione del glucosio standard nella cura del diabete: un beneficio per la salute cognitiva “, afferma la prima autrice, la professoressa Katherine Samaras, leader del tema di ricerca sull’invecchiamento sano presso il Garvan Institute ed endocrinologa presso il St Vincent’s Ospedale Sydney.
Protezione della funzione cerebrale
Il diabete di tipo 2 si verifica quando il corpo non è più in grado di produrre abbastanza insulina per soddisfare i suoi bisogni, lasciando le persone colpite incapaci di mantenere i livelli di glucosio nel sangue entro un intervallo normale.
Ciò può portare a complicazioni di salute a lungo termine, incluso il declino cognitivo.
“Con l’avanzare dell’età, le persone che convivono con il diabete di tipo 2 hanno un incredibile rischio del 60% di sviluppare demenza, una condizione devastante che influisce sul pensiero, sul comportamento, sulla capacità di svolgere le attività quotidiane e sulla capacità di mantenere l’indipendenza.
Ciò ha enormi impatti personali, familiari, economici e sociali “, afferma il professor Samaras.
I ricercatori di questo studio hanno esaminato i dati dei partecipanti al Sydney Memory and Aging Study di CHeBA. In questa coorte, 123 partecipanti allo studio avevano il diabete di tipo 2 e 67 hanno ricevuto metformina per abbassare i livelli di zucchero nel sangue.
I ricercatori hanno testato la funzione cognitiva ogni due anni, utilizzando valutazioni dettagliate che misuravano la cognizione su una serie di capacità, tra cui memoria, funzione esecutiva, attenzione e velocità e linguaggio.
I risultati hanno rivelato che gli individui con diabete di tipo 2 che assumevano metformina avevano un declino cognitivo significativamente più lento e un rischio di demenza inferiore rispetto a quelli che non assumevano metformina.
Sorprendentemente, in quelli con diabete di tipo 2 che assumevano metformina , non c’era alcuna differenza nel tasso di declino della funzione cognitiva in 6 anni rispetto a quelli senza diabete.
Nuovo utilizzo per un comune farmaco
La metformina è stata utilizzata in modo sicuro per il trattamento del diabete di tipo 2 per 60 anni. Agisce riducendo la quantità di glucosio rilasciato dal fegato nel flusso sanguigno e consente alle cellule del corpo di rispondere meglio ai livelli di glucosio nel sangue.
Studi dell’ultimo decennio hanno rivelato prove del beneficio della metformina nel cancro, malattie cardiache, sindrome dell’ovaio policistico e gestione del peso.
Mentre l’attuale studio suggerisce che la metformina può avere benefici cognitivi per le persone che convivono con il diabete di tipo 2, i ricercatori affermano che potrebbe anche giovare a chi è a rischio di declino cognitivo più in generale.
“Questo studio ha fornito una promettente evidenza iniziale che la metformina può proteggere dal declino cognitivo.
Mentre si ritiene che il diabete di tipo 2 aumenti il rischio di demenza promuovendo percorsi degenerativi nel cervello e nei nervi, questi percorsi si verificano anche in altri a rischio di demenza ed è possibile che l’insulino-resistenza possa essere il mediatore “, afferma il professor Samaras.
“Per stabilire un effetto definitivo, stiamo ora pianificando un ampio studio controllato randomizzato di metformina in soggetti a rischio di demenza e valutiamo la loro funzione cognitiva per tre anni. Questo potrebbe tradursi nella nostra capacità di riutilizzare questo farmaco economico con un robusto profilo di sicurezza per aiutare a prevenire il declino cognitivo nelle persone anziane “.
Il Sydney Memory and Aging Study del CHeBA è uno studio osservazionale degli australiani più anziani che è iniziato nel 2005 e ricerca gli effetti dell’invecchiamento sulla cognizione nel tempo.
Il professor Perminder Sachdev, autore senior dello studio e co-direttore del CHeBA, afferma: “Sebbene uno studio osservazionale non fornisca una ‘prova’ conclusiva che la metformina sia protettiva contro la demenza, ci incoraggia a studiare questo e altri trattamenti antidiabetici per la prevenzione della demenza.
È stato persino suggerito che la metformina sia antietà .
La domanda interessante è se la metformina sia utile nelle persone con normale metabolismo del glucosio. È chiaramente necessario più lavoro. “
MA…………
La metformina è tra i farmaci più frequentemente prescritti in tutto il mondo ed è utilizzata per facilitare il catabolismo del glucosio nei pazienti con ridotta segnalazione dell’insulina (diabete di tipo 2) (Salani et al., 2014).
Si ritiene che la metformina agisca inibendo la respirazione mitocondriale (Wheaton et al., 2014). Recentemente, la metformina è stata testata per ulteriori effetti fisiologici e si è scoperto che prolunga la durata della vita in modelli animali che vanno dai nematodi ai topi (Martin-Montalvo et al., 2013; Onken e Driscoll, 2010).
L’ampio uso clinico nell’uomo ha consentito la raccolta e l’analisi dei dati sulla longevità dei pazienti affetti da diabete trattati con metformina. La coorte di diabete esposta alla metformina ha una vita più lunga rispetto ai soggetti sani non trattati (Bannister et al., 2014), in linea con le potenziali proprietà di prolungamento della vita della metformina.
Il diabete di tipo 2 è un disturbo associato all’invecchiamento e molti pazienti iniziano il trattamento con metformina in tarda età. Sulla base dell’analisi di sopravvivenza dei pazienti diabetici, è stato proposto che gli effetti di prolungamento della vita della metformina possano estendersi anche a soggetti anziani sani dal punto di vista metabolico.
Considerando gli effetti collaterali moderati della metformina nel diabete e i potenziali benefici dell’invecchiamento in buona salute, la metformina è emersa come un candidato interessante per essere testato clinicamente come il primo potenziale farmaco antietà negli esseri umani.
Recentemente è stato completato uno studio a breve termine sulla somministrazione di metformina a soggetti pre-diabetici di età (≥65 anni) per un periodo di 6 settimane e sono stati riportati i dati (Kulkarni et al., 2018). Mentre sono stati osservati effetti su percorsi come TOR e risposta immunitaria, non sono stati rilevati evidenti cambiamenti fisiologici a causa della breve durata del trattamento, lasciando una questione aperta agli effetti a lungo termine della metformina su esseri umani sani anziani.
Questa domanda è tuttavia fondamentale perché si prevede che gli anziani non diabetici saranno i primi destinatari del presunto trattamento di estensione della durata della salute con metformina.
Attraverso la ricerca della letteratura su studi sugli animali che forniscono prove della modulazione della longevità da parte della metformina, abbiamo scoperto che l’effetto pro-sopravvivenza di questo farmaco è stato studiato principalmente in animali giovani o animali esposti a metformina dalla giovane età adulta.
I pochi studi condotti su animali più anziani (topi di 56-60 settimane di età, vita media 96 settimane; nematodi di 8 giorni, durata media di vita 14-21 giorni), non sono riusciti a rilevare l’estensione della vita con la metformina (Alfaras et al., 2017; Anisimov et al., 2011) o ha rivelato una tossicità parzialmente attribuita al sovradosaggio di metformina (Cabreiro et al., 2013; Martin-Montalvo et al., 2013; Thangthaeng et al., 2017).
Sorprendentemente, una dose di 50 mM di metformina che ha innescato il più forte prolungamento della durata della vita in uno studio fondamentale condotto su giovani C. elegans, era moderatamente tossica quando somministrata a nematodi di mezza età (età adulta giorno 8) (Cabreiro et al., 2013; Onken e Driscoll, 2010 ).
Siamo quindi giunti alla conclusione che i benefici e la sicurezza della somministrazione di metformina a soggetti anziani non resistenti all’insulina non erano stati sufficientemente studiati, contrariamente alle risposte dei pazienti diabetici.
Qui abbiamo utilizzato test genetici, misurazioni metaboliche, test reporter dello stress e analisi omiche per rilevare gli effetti specifici dell’età della metformina in C. elegans e nelle cellule primarie umane. Abbiamo scoperto che il trattamento con metformina iniziato in tarda età accorcia la durata della vita e limita la sopravvivenza cellulare aggravando la disfunzione mitocondriale associata all’invecchiamento verso l’insufficienza respiratoria.
Oltre alla distorsione mitocondriale, le vecchie cellule non sono riuscite a migliorare l’uso della glicolisi in risposta alla metformina, portando ad un persistente esaurimento dell’ATP. Abbiamo scoperto che gli interventi che stabilizzano i livelli di ATP cellulare, come la replezione di ATP e l’inibitore TOR rapamicina, alleviano la tossicità della metformina in età avanzata in vitro e in vivo.
Abbiamo anche scoperto che il trattamento con metformina in età precoce istiga una serie di stress e adattamenti metabolici che probabilmente sono alla base dell’estensione della longevità da parte della metformina. È importante sottolineare che l’induzione di queste risposte favorevoli è stata fortemente compromessa nella tarda età.
In particolare, dimostriamo che il trattamento con metformina precoce ma non tardiva induce una risposta del turnover lipidico simile alla restrizione dietetica (DR). Abbiamo anche scoperto che questo fenotipo mimetico DR della prima infanzia è istigato dalla via della lipolisi dei trigliceridi regolata dalla proteina chinasi A e non dalla via della proteina chinasi attivata da AMP (AMPK), come suggerito in precedenza.
Successivamente, abbiamo dimostrato che il trattamento con metformina limitato alla prima età adulta è sufficiente per l’estensione della vita, rafforzando il ruolo chiave dello stress precoce della vita e degli adattamenti metabolici nei benefici della metformina sulla longevità. Infine, dimostriamo che i mutanti daf-2 (e1370), portatori di un’insufficienza simile al diabete del recettore dell’insulina C. elegans, sono resistenti alla tossicità della metformina in età avanzata rispetto ai controlli wild type di pari età, grazie alla migliore capacità di sostenere l’ATP sintesi durante l’esposizione alla metformina in età avanzata. Collettivamente, abbiamo scoperto un’allarmante capacità della metformina di indurre un fallimento metabolico in soggetti anziani non insulino-resistenti, che potrebbe limitare i suoi benefici per gli anziani non diabetici.
Risultati
Il trattamento con metformina nella tarda età è dannoso per la longevità
Per affrontare i risultati del trattamento con metformina a diverse età, abbiamo trattato il giovane adulto (3 giorni di età, giorno 1 dell’età adulta), l’adulto all’età del declino della riproduzione (giorno 4 dell’età adulta), metà di età (giorno 8 dell’età adulta) e vecchi (giorno 10 dell’età adulta) di C. elegans di tipo selvatico con diverse dosi di metformina – 10 mM, 25 mM e 50 mM. La metformina 50 mM è la dose comune utilizzata per indurre l’estensione della durata della vita in C. elegans mentre 10 mM è la dose più bassa legata all’estensione della vita riproducibile in questo modello in rapporti precedenti (Cabreiro et al., 2013; Onken e Driscoll, 2010; Pryor et al. , 2019).
Abbiamo scoperto che il trattamento con metformina è iniziato in giovane età (giorni 1 e 4 dell’età adulta) ha prolungato la durata della vita dei nematodi a tutte le dosi utilizzate (Figura 1A e Figura S1A).
All’interno del trattamento iniziato il giorno 8 dell’età adulta, le dosi di 50 mM e 25 mM di metformina hanno ridotto la durata della vita mediana ma hanno prolungato la durata massima in linea con le osservazioni precedenti (Cabreiro et al., 2013) mentre la dose di 10 mM era prolungata dalla longevità senza effetti dannosi (Figura S1B) .
Sorprendentemente, il giorno 10 dell’età adulta la metformina era tossica a tutte le dosi utilizzate con un’ampia percentuale di animali esposti al farmaco che morivano entro le prime 24 ore dal trattamento (Figura 1B). I nostri primi esperimenti sui nematodi hanno quindi rivelato un’evidente diminuzione dipendente dall’età della tolleranza alla metformina che è culminata nella tossicità in tarda età di tutte le dosi di metformina testate, indicando possibili rischi per la sicurezza della somministrazione di metformina in tarda età.

trattamento con metformina iniziato in tarda età esercita tossicità e limita la sopravvivenza indipendentemente da AMPK e cambiamenti microbici.
I nematodi wild type (WT, ceppo N2 Bristol) sono stati trattati con dosi indicate di metformina (Met) il giorno 1 (A) e il giorno 10 (B) dell’età adulta (AD1 e AD10 rispettivamente), la sopravvivenza è stata valutata giornalmente. I nematodi WT sono stati coltivati su E. coli HT115 (C) e OP50 (D) vivi e uccisi con UV e trattati con metformina 50 mM su AD10. La sopravvivenza è stata valutata giornalmente. I nematodi WT (E, a sinistra) e con deficit di AMPK (E, a destra) sono stati trattati con metformina 50 mM su AD1 e AD10, la sopravvivenza è stata valutata giornalmente. La significatività è stata misurata mediante test log-rank, n≥100 in tutti i casi, gli esatti n numeri e valori statistici per tutti i pannelli sono presentati nella Tabella S1. Ogni esperimento è stato ripetuto ≥3 volte; in tutti i casi viene mostrato un risultato rappresentativo.
La tossicità della metformina nella tarda età è indipendente dal microbioma
Per comprendere il meccanismo della tossicità della metformina nella tarda età, abbiamo prima affrontato i percorsi noti che regolano l’estensione della durata della vita di questo composto. L’effetto pro-longevità della metformina nei giovani nematodi di C. elegans era precedentemente collegato ai cambiamenti del metabolismo microbico indotti da questo farmaco (Cabreiro et al., 2013).
Per verificare se la tossicità della metformina in età avanzata si basava su alterazioni del microbioma simili, abbiamo trattato i vermi con metformina in presenza di ceppi di E. coli OP50 (metformina sensibili) e HT115 (resistenti alla metformina) vivi e uccisi dai raggi UV.
La tossicità della metformina nell’età avanzata si è sviluppata indipendentemente dalla vitalità batterica e / o dal ceppo (Figura 1C e D) suggerendo che l’intolleranza alla metformina nella tarda età è indipendente dai cambiamenti del microbioma precedentemente scoperti. È interessante notare che la sopravvivenza di base dei nematodi differiva tra le diete OP50 e HT115 uccise dai raggi UV in linea con la dipendenza recentemente segnalata dei comportamenti fisiologici dei nematodi dalla fonte batterica (Revtovich et al., 2019).
L’AMPK non è richiesto per la tossicità della metformina
in età avanzata Un altro componente essenziale per l’effetto di prolungamento della vita della metformina in giovane età è la protein chinasi attivata da AMP (AMPK); in particolare la metformina non è riuscita a promuovere la longevità nei nematodi privi dell’ortologo AMPK AAK-2 (Onken e Driscoll, 2010). Al fine di sondare il requisito di AMPK per la tossicità della metformina in età avanzata, abbiamo trattato animali mutanti giovani e anziani di tipo selvatico e aak-2 (ok524) con questo farmaco. Coerentemente con i rapporti precedenti, il trattamento con metformina nella prima infanzia non è stato in grado di indurre l’estensione della durata della vita nei vermi carenti di aak-2 (Figura 1E); allo stesso tempo la tossicità della metformina in età avanzata si è sviluppata negli animali mutanti, indicando che l’accorciamento della vita indotto dalla metformina in età avanzata non viene eseguito dall’AMPK.
La tossicità della metformina è innescata da alterazioni mitocondriali
Una delle funzioni primarie della metformina è quella di inibire il complesso I della catena di trasporto degli elettroni mitocondriali (ETC), che influenza il potenziale della membrana mitocondriale e la produzione di ATP (Andrzejewski et al., 2014; Cameron et al., 2018; Wheaton et al., 2014).
L’inibizione della crescita da parte della metformina era precedentemente collegata a una respirazione mitocondriale ridotta nei nematodi e nelle cellule di mammifero (Wu et al., 2016). Inoltre, l’accumulo di mitocondri danneggiati e disfunzionali, che può aumentare l’impatto negativo degli inibitori del complesso ETC I sulla sopravvivenza cellulare, è uno dei segni distintivi dell’invecchiamento meglio caratterizzati (Bratic e Larsson, 2013; Bratic e Trifunovic, 2010; Cellerino e Ori , 2017; Sun et al., 2016; Taylor e Dillin, 2011).
Il deterioramento mitocondriale paragonabile all’invecchiamento si verifica prematuramente negli animali mutanti, difettoso nella biogenesi mitocondriale e nel controllo di qualità (Sun et al., 2016; Trifunovic et al., 2004). Per verificare se la tossicità della metformina durante l’invecchiamento (e la tossicità della metformina in generale) è guidata dall’accumulo di menomazioni mitocondriali, abbiamo ottenuto mutanti che ospitano carenze di diverse vie dell’omeostasi mitocondriale: risposta della proteina spiegata mitocondriale (atfs-1 (gk3094)) (Nargund et al. , 2012), biogenesi mitocondriale (skn-1 (zj15)) (Palikaras et al., 2015), respirazione mitocondriale (isp-1 (qm150)) (Feng et al., 2001) e controllo della qualità delle proteine mitocondriali (ubl-5 (ok3389)) (Benedetti et al., 2006) e hanno trattato questi animali con metformina insieme a controparti selvatiche.
Una combinazione di metformina con alterazioni mitocondriali congenite ha portato a un esordio precoce della tossicità da metformina in tutti i background mutanti testati (compresi mutanti isp-1 (qm150) normalmente a vita lunga) che collegano chiaramente l’intolleranza alla metformina all’elevata abbondanza di mitocondri disfunzionali (Figura 2A- C, Figura S2A).
Lo stesso effetto (insorgenza prematura della tossicità da metformina in giovane età) è stato osservato nei nematodi e nelle cellule primarie umane incubate con l’agente di disaccoppiamento mitocondriale carbonil cianuro-p-trifluorometossifenilidrazone (FCCP) insieme alla somministrazione di metformina (Figura 2D-E e Figura S2B).
Da notare che recentemente è stato scoperto che i difetti congeniti della respirazione mitocondriale limitano l’interazione che prolunga la vita tra metformina e microbioma (Pryor et al., 2019) in linea con il ruolo chiave dell’integrità mitocondriale nei diversi benefici per la longevità della metformina.
Per verificare se la mitofagia e / o la risposta proteica dispiegata mitocondriale (UPR MT) – le vie protettive che rispondono al fallimento mitocondriale e note per deteriorarsi durante l’invecchiamento (Sun et al., 2016), sono state indotte direttamente dalla metformina, abbiamo misurato l’abbondanza di proteine mitocondriali e l’espressione del transgene hsp-6 :: gfp (reporter UPR MT) in animali giovani e anziani trattati con questo farmaco.
A entrambe le età la somministrazione di metformina non ha portato né a un’elevata espressione di GFP né a una riduzione dei livelli di proteina mitocondriale (Figura S3A-C), indicando che la mitofagia e la MT UPR non sono innescate in modo prominente dalla metformina e probabilmente non svolgono alcun ruolo nell’adattamento cellulare immediato a effetti indotti dalla metformina.
Da notare, la mancanza di induzione della MT UPR da parte della metformina è stata precedentemente segnalata da un gruppo indipendente (De Haes et al., 2014). Mostriamo quindi che l’inizio precoce della tossicità della metformina nei mutanti mitocondriali è probabilmente determinato dall’accumulo di danni mitocondriali prima della somministrazione di metformina, paragonabile a ciò che si verifica durante l’invecchiamento.

La tossicità della metformina si associa all’esaurimento dell’ATP ed è alleviata dalla replezione dell’ATP
Per misurare gli effetti diretti specifici dell’età della metformina sulle prestazioni mitocondriali e per testare la conservazione dei nostri risultati nell’uomo, abbiamo analizzato l’impatto del trattamento con metformina sull’omeostasi del passaggio precoce (giovani) e fibroblasti primari della pelle umana a passaggio tardivo (vecchi, senescenti replicativi).
Il modello di senescenza replicativa è stato scelto per la sua elevata rilevanza per il normale invecchiamento umano poiché le cellule senescenti si accumulano nei tessuti che invecchiano. L’invecchiamento in vitro è stato eseguito secondo procedure standard che hanno dimostrato di produrre cellule portatrici dei principali segni distintivi dell’invecchiamento (Tigges et al., 2014) e il declino mitocondriale associato all’invecchiamento delle cellule tardive è stato verificato dall’analisi Seahorse (Figura 3C-D, Figura S4A e C).
Abbiamo scoperto che, a dosi elevate, la metformina era tossica sia per le cellule giovani che per quelle vecchie, ma le cellule vecchie (simili ai vecchi nematodi) mostravano un declino della vitalità molto più forte e soccombevano alla tossicità già a dosi più basse di metformina (Figura 3A-B).
Le misurazioni del tasso di consumo di ossigeno (OCR) hanno dimostrato un effetto significativo della metformina sulla respirazione basale nelle cellule giovani e vecchie (Figura 3C, Figura S4A), mentre l’inibizione della sintesi di ATP mitocondriale da parte della metformina era più forte nei vecchi fibroblasti (Figura S4C).
Inoltre, solo le cellule vecchie hanno mostrato una diminuzione della respirazione massima in risposta alla metformina (Figura 3D, Figura S4A) coerente con un impatto negativo più forte di questo farmaco sui mitocondri delle cellule vecchie. È interessante notare che il tasso di acidificazione extracellulare (ECAR) è stato notevolmente aumentato nei fibroblasti giovani trattati con metformina (Figura 3E, Figura S4B) in linea con la loro elevata dipendenza dalla glicolisi in risposta all’insufficienza mitocondriale innescata dalla metformina.
Questo aumento adattativo della glicolisi è stato notevolmente ridotto nelle vecchie cellule esposte alla metformina (Figura 3E, Figura S4B), privando queste cellule, in combinazione con il più forte ostacolo mitocondriale della metformina, di percorsi efficaci della sintesi di ATP. La successiva determinazione del contenuto di ATP cellulare ha infatti mostrato un forte calo dei livelli di ATP nelle vecchie cellule trattate con metformina (Figura 3F).
Abbiamo anche rilevato una più forte distorsione del potenziale di membrana mitocondriale nei fibroblasti esposti alla metformina vecchi rispetto a quelli giovani (Figura 3G), in linea con un declino mitocondriale più potente osservato nelle cellule vecchie tramite analisi del consumo di ossigeno.

Successivamente abbiamo misurato l’effetto della metformina sul contenuto di ATP dell’organismo nei nematodi. Il trattamento con metformina di animali giovani non ha causato cambiamenti negativi dei livelli sistemici di ATP (Figura 3H), coerenti con ridotte menomazioni mitocondriali e intatta adattabilità metabolica in giovane età.
Sorprendentemente, gli animali anziani hanno mostrato una forte riduzione dei livelli di ATP di base, rispetto agli animali giovani non trattati, seguita da un ulteriore calo del 76% del contenuto di ATP indotto dalla metformina (Figura 3H, Tabella S2). Questi risultati sono coerenti con il noto deterioramento mitocondriale e con la riduzione dell’energetica di vecchi animali (e cellule) (Brys et al., 2010; Drew et al., 2003) e suggeriscono, insieme ai fenotipi metabolici specifici dell’età sopra riportati, che la tossicità della metformina può essere collegato a un fallimento delle vecchie cellule nel mantenere la sintesi di ATP durante il trattamento con metformina, portando a una diminuzione del contenuto di ATP fino a livelli incompatibili con la vitalità cellulare.
Per testare questa ipotesi abbiamo chiesto se l’integrazione ectopica di ATP avrebbe salvato la tossicità da metformina. Fornire ATP agli animali non è fattibile a causa della biodisponibilità insufficiente dopo l’integrazione orale (Arts et al., 2012), siamo stati tuttavia in grado di integrare l’ATP ai fibroblasti in coltura utilizzando concentrazioni millimolari come descritto in precedenza (1978).
La replezione di ATP ectopica ha alleviato la morte cellulare, l’esaurimento dell’ATP e la perdita del potenziale di membrana mitocondriale indotto dalla metformina (Figura 3I-J, Figura S5A-D) e da un altro rotenone inibitore del complesso I (Figura S6A-C) che collega la perdita di vitalità al trattamento con il complesso ETC Inibitori delle alterazioni dell’omeostasi energetica.
Il salvataggio del potenziale di membrana mitocondriale mediante la replezione di ATP era coerente con la capacità precedentemente riportata dell’ATP sintasi mitocondriale di ripristinare il potenziale di membrana in condizioni di insufficienza respiratoria consumando ATP (Chinopoulos e Adam-Vizi, 2010).
La tossicità della metformina è sospesa nei nematodi portatori di insufficienza del recettore dell’insulina
Poiché l’intolleranza alla metformina, paragonabile ai nostri risultati, non è stata segnalata nei pazienti con diabete di tipo 2 che ricevevano un trattamento in tarda età, abbiamo deciso di testare la risposta alla metformina di vecchiaia dei nematodi privi di recettore dell’insulina funzionale ( un mimetico molecolare della resistenza all’insulina nel diabete di tipo 2).
Per questo abbiamo scelto un ceppo daf-2 (e1370) ben caratterizzato che porta una mutazione sensibile alla temperatura dell’ortologo DAF-2 del recettore C. elegans IGF-1 / insulina (Dorman et al., 1995). A una temperatura restrittiva di 20 ° C, i mutanti daf-2 (e1370) mostrano una perdita parziale della funzione del recettore dell’insulina che porta a una varietà di fenotipi, parzialmente paragonabili ai pazienti diabetici (come un aumento della deposizione di grasso) (Kimura et al., 1997; Tissenbaum e Ruvkun, 1998).
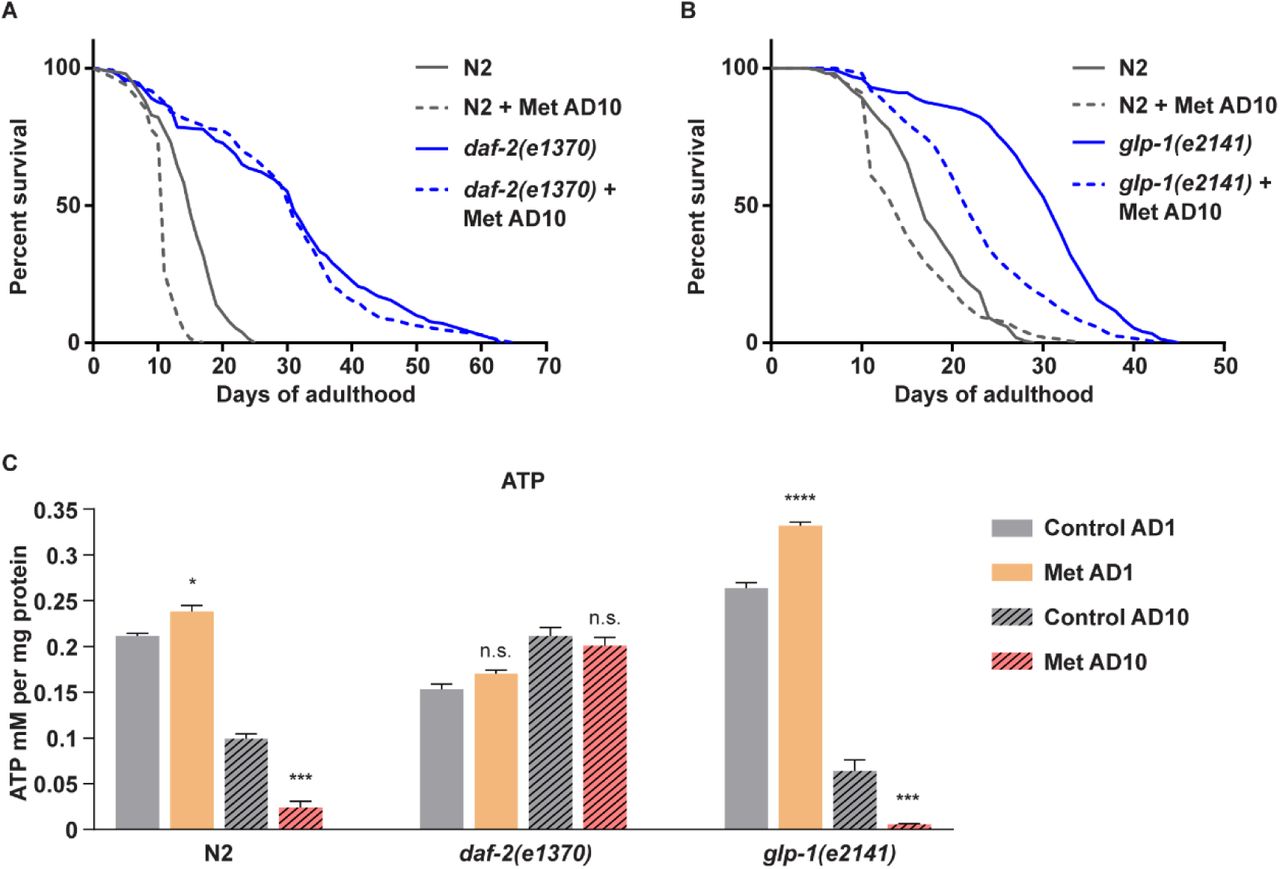
Successivamente abbiamo misurato la sopravvivenza dei daf-2 (e1370) e dei nematodi selvatici trattati con metformina il giorno 10 dell’età adulta (AD10) e abbiamo osservato una sorprendente resilienza dei mutanti all’uccisione della metformina a questa età (Figura 4A). Le misurazioni sistemiche di ATP hanno rivelato una moderata riduzione della sintesi di ATP al basale nei giovani mutanti daf-2 (e1370) (Figura 4C) coerente con i rapporti precedenti (Brys et al., 2010; Palikaras et al., 2015).
È interessante notare che né l’invecchiamento né la metformina hanno avuto un impatto negativo sui livelli di ATP negli animali AD10 daf-2 (e1370), contrariamente alla risposta dei controlli di tipo selvatico (Figura 4C), associando la resilienza dei mutanti alla tossicità della metformina con la loro capacità di sostenere l’ATP sintesi durante il trattamento con metformina in tarda età. Da notare, la maggiore competenza dei mutanti daf-2 (e1370) nella sintesi di ATP durante la tarda età è anche coerente con i rapporti precedenti (Brys et al., 2010; Palikaras et al., 2015).
È importante sottolineare che la resistenza degli animali daf-2 (e1370) alla metformina non era infinita e i nematodi mutanti esposti al farmaco il giorno 21 dell’età adulta (AD21) hanno mostrato livelli di tossicità da metformina paragonabili ai controlli di tipo selvatico AD10 (Figura S7A), indicando che il recettore dell’insulina stesso non è necessario per l’induzione della tossicità della metformina in età avanzata.
In sintesi, questi dati ribadiscono il ruolo chiave dell’esaurimento dell’ATP nella tossicità della metformina in tarda età e indicano che l’inibizione della segnalazione del recettore dell’insulina, paragonabile all’insulino-resistenza nel diabete, fornisce una protezione estesa dall’intolleranza alla metformina in età avanzata.
La resilienza della metformina dei mutanti del recettore dell’insulina non è dovuta al loro tasso di invecchiamento più lento
I mutanti daf-2 (e1370) sono noti per essere longevi rispetto agli animali selvatici (Dorman et al., 1995; Kimura et al., 1997 ; Tissenbaum e Ruvkun, 1998). Per garantire che la resilienza prolungata alla metformina degli animali daf-2 (e1370) non sia una semplice conseguenza del loro più lento tasso di invecchiamento, abbiamo selezionato un diverso ceppo di lunga durata: mutanti glp-1 (e2141) privati della linea germinale (Lapierre et al., 2011) e hanno trattato questi animali con metformina il giorno 10 dell’età adulta (AD10).
A differenza dei nematodi daf-2 (e1370), i vermi glp-1 (e2141) hanno ceduto all’uccisione della metformina AD10 simile ai controlli wild type (Figura 4B). Hanno anche mostrato cali sia innescati dall’invecchiamento che aggravati dalla metformina dei livelli di ATP, a differenza dei mutanti AD10 daf-2 (e1370) e simili ai controlli wild type (Figura 4C).
Abbiamo così determinato che la capacità di sostenere la sintesi di ATP in tarda età e la corrispondente resilienza della metformina non sono fenotipi comuni di tutti i mutanti di lunga durata, ma piuttosto una caratteristica specifica degli animali con deficit del recettore dell’insulina.
La resilienza della metformina dei mutanti del recettore dell’insulina dipende dalla sintesi prolungata di ATP in tarda età regolata da DAF-16 / FOXO.
L’esatto meccanismo alla base della maggiore competenza metabolica dei mutanti daf-2 (e1370) in tarda età resta da chiarire. Tuttavia, simile alla maggior parte dei fenotipi benefici daf-2 (e1370), questa capacità può dipendere dal fattore di trascrizione FOXO DAF-16 (Kenyon, 2011; Palikaras et al., 2015). Coerentemente, daf-2 (e1370); I doppi mutanti daf-16 (mu86) non hanno mostrato resilienza alla metformina nel giorno 10 dell’età adulta, a differenza dei mutanti singoli daf-2 (e1370) e simili ai controlli wild-type (Figura S7B).
I doppi mutanti hanno anche mostrato un chiaro declino della sintesi di ATP dopo il trattamento con metformina AD10 (Figura S7C) in linea con il requisito di DAF-16 per la resilienza metabolica del background genetico daf-2 (e1370). Poiché DAF-16 è un importante mediatore di adattamenti multipli allo stress (Kenyon, 2011), abbiamo testato se la risposta alla metformina richiedesse DAF-16 direttamente misurando la traslocazione nucleare della proteina di fusione DAF-16 :: GFP durante il trattamento con metformina o shock termico (usato come controllo positivo).
Mentre DAF-16 si stava chiaramente traslocando nel nucleo in risposta allo stress da calore, è stata osservata una piccola traslocazione (al di sopra dei valori di controllo negativi) nel corso del trattamento con metformina indipendentemente dall’età (Figura S8A-C), suggerendo che la resilienza della metformina di daf- 2 (e1370) mutanti non sono guidati dal coinvolgimento diretto di DAF-16 negli adattamenti dello stress durante il trattamento con metformina.
La capacità degli animali daf-2 (e1370) di sostenere una sintesi stabile di ATP in età avanzata è stata precedentemente collegata a un elevato turnover mitocondriale indotto dallo stress metabolico dei sottotitoli per tutta la vita, che porta a una migliore qualità mitocondriale nella tarda età (Brys et al., 2010 ; Palikaras et al., 2015). Le nostre misurazioni di proteomica hanno infatti rivelato livelli basali ridotti di proteine mitocondriali nei mutanti daf-2 (e1370) rispetto agli animali selvatici (Figura S7D) coerenti con un elevato turnover mitocondriale.
È interessante notare che i livelli di proteina mitocondriale di daf-2 (e1370); I doppi mutanti daf-16 (mu86) erano simili ai valori osservati negli animali selvatici (Figura S7D), suggerendo che il reintegro mitocondriale innescato dal deficit del recettore dell’insulina è inibito in assenza di DAF-16.
Coerentemente con il ruolo chiave di DAF-16 nel supportare l’omeostasi metabolica dei mutanti daf-2 (e1370), il daf-2 (e1370); I doppi mutanti daf-16 (mu86) hanno dimostrato forti alterazioni della sintesi di ATP di base già in giovane età (Figura S7C).
La metformina attiva rapidamente diversi percorsi di garanzia della longevità in animali giovani ma non anziani
Per verificare se altre risposte di metformina, oltre alla sintesi di ATP, vengono alterate in tarda età, abbiamo eseguito un’analisi proteomica imparziale degli animali trattati con metformina nei giorni 1 e 10 dell’età adulta per 24 e 48 ore e confrontati con i controlli non trattati con corrispondenza per età e punto temporale.
L’abbondanza di solo una percentuale limitata di gruppi proteici è stata influenzata dalla metformina ad entrambe le età e in entrambi i momenti del trattamento (osservata confrontando il numero di proteine alterate (cerchiate) e di proteine non alterate (riquadro esterno) nelle Figure 5A e S9A), evidenziando la specificità della risposta suscitata dal trattamento farmacologico.
Confrontando i profili di espressione proteica ottenuti (4572 gruppi proteici sono stati quantificati in totale, Tabella S3), abbiamo determinato che le risposte globali alla metformina in giovane e vecchiaia erano ampiamente distinte, come indicato da una limitata sovrapposizione tra le proteine comunemente colpite sia a 24 che a 48 ore di trattamento (Figura 5A e S9A) e una modesta correlazione tra i cambiamenti proteici indotti (Figura 5B e S9B).
È interessante notare che la somiglianza tra le risposte di giovani e anziani era più forte a 48 ore di trattamento, il che potrebbe essere spiegato da un ritardo degli animali anziani nella risposta al farmaco o da un arricchimento del campione di 48 ore di vecchiaia esposto alla metformina con animali che erano più competenti nel loro adattamento al trattamento con metformina (sulla base delle nostre analisi di sopravvivenza una percentuale significativa di animali anziani sensibili alla metformina è morta tra le 24 e le 48 ore di trattamento).

Analizzando i percorsi regolati dalla metformina in punti di età distinti, abbiamo rilevato una rapida attivazione delle risposte adattive allo stress e dei percorsi di assicurazione della longevità come la sottoregolazione del ribosoma (MacInnes, 2016) e l’induzione della risposta allo stress ossidativo (Honda e Honda, 1999), risposta (Xia et al., 2019) e risposta allo stress da calore (Baldi et al., 2017) in animali giovani (Figura 5C-E e S9C). Anche l’autofagia generale (Hansen et al., 2018) è stata regolata in modo comparabile, sebbene in misura minore (Figura S9D).
Questi dati sono coerenti con studi precedenti che suggeriscono che la metformina prolunga la durata della vita inducendo adattamenti allo stress come la risposta allo stress ossidativo (De Haes et al., 2014; Onken e Driscoll, 2010).
A differenza dei rapporti precedenti, tuttavia, i nostri dati dimostrano per la prima volta che la metformina innesca simultaneamente una serie complessa di diverse risposte allo stress che probabilmente contribuiscono tutte all’estensione della vita di questo farmaco.
Sorprendentemente, l’induzione degli adattamenti allo stress da parte della metformina è stata inibita o ritardata negli animali anziani (Figura 5C-E, Figura S9C-D) suggerendo che, oltre all’esaurimento dell’ATP innescato dalla metformina, anche la componente di garanzia della longevità della risposta alla metformina è compromessa in tarda età.
Per studiare la specificità dell’età delle risposte allo stress innescate dalla metformina con un metodo indipendente, abbiamo misurato l’induzione dell’autofagia generale da parte della metformina in un ceppo transgenico che esprime la proteina di fusione LGG-1 :: mCherry (Gosai et al., 2010).
LGG-1 è un ortologo LC3 di C. elegans che viene reclutato nelle membrane autofagosomiche durante il processo di autofagia, dando origine (in un ambiente reporter) a punti distinti che possono essere quantificati microscopicamente (Figura S10A).
L’autofagia è stata scelta per il test di convalida a causa del suo noto effetto antagonista sulla longevità a seconda dell’età (Wilhelm et al., 2017) e dell’integrità mitocondriale (Zhou et al., 2019).
Trattando animali transgenici giovani e anziani con metformina, abbiamo potuto effettivamente osservare una sostanziale induzione della formazione di autofagosomi in animali esposti a metformina giovani ma non anziani (Figura S10B-C) con dimensioni degli effetti paragonabili ai dati della proteomica (Figura S9D).
È interessante notare che il numero assoluto di autofagia puncta negli animali di controllo non trattati era significativamente più alto in età avanzata (Figura S10D-E) coerente con l’inibizione precedentemente osservata del flusso di autofagia durante la tarda età (Wilhelm et al., 2017).
La specificità del puncta osservato per il processo di autofagia è stata confermata dall’esposizione di animali transgenici all’RNAi contro il mediatore chiave dell’autofagia beclin 1 (bec-1) (Figura S10F).
In sintesi, abbiamo scoperto che il trattamento con metformina esercita una serie di risposte di adattamento allo stress negli animali giovani mentre gli animali anziani non riescono ad attivare questi segnali in misura comparabile in linea con precedenti accenni di ridotta resilienza allo stress in età avanzata (Haigis e Yankner, 2010).
Per sondare il contributo degli eventi di adattamento della prima infanzia all’effetto pro-longevità della metformina, abbiamo esposto i nematodi al farmaco durante la prima età adulta e in effetti abbiamo scoperto che il trattamento con metformina limitato alla giovane età è sufficiente per conferire benefici in vivo sulla durata della vita (Figura S10G).
La risposta alla metformina specifica per la vecchiaia è arricchita in mediatori distinti del metabolismo dei lipidi
. Successivamente abbiamo esaminato i dati di proteomica per le vie attivate dalla metformina prevalentemente negli animali anziani. Abbiamo scoperto che i componenti perossisomiali insieme agli enzimi mitocondriali e perossisomiali implicati nella β-ossidazione degli acidi grassi (come acs-1, acs-2, acox-2) (Zhang et al., 2011) erano sovraregolati nei vecchi più forti che nei giovani animali (Figura 5F, H).
Anche le proteine associate alle goccioline di lipidi, che servono come unità chiave di immagazzinamento dei lipidi in C. elegans, (vitellogenine, deidrogenasi) (Vrablik et al., 2015; Zhang et al., 2012) erano sovraregolate nei vecchi mentre erano sottoregolate nei giovani trattati con metformina animali (Figura 5G, Figura S9E).
Inoltre, i componenti ribosomiali trovati per essere parte del proteoma delle goccioline lipidiche (Vrablik et al., 2015; Zhang et al., 2012) erano sottoregolati nei nematodi esposti alla metformina giovani ma non nei vecchi (Figura S9C). Collettivamente, i nostri dati hanno indicato che il trattamento con metformina porta ad un aumento del contenuto perossisomiale, che è più forte all’età avanzata rispetto alla giovane età, mentre i componenti delle goccioline lipidiche sono sottoregolati nei giovani e sovraregolati negli animali anziani.
È interessante notare che la sottoregolazione delle goccioline lipidiche e la sovraregolazione dei perossisomi da parte della metformina negli animali giovani sono state recentemente segnalate da uno studio indipendente (Pryor et al., 2019) che tuttavia non ha affrontato la specificità dell’età di questi fenotipi.
Il trattamento con metformina porta a risposte distinte del turnover lipidico negli animali giovani e anziani
Poiché l’analisi proteomica ha rivelato la regolazione specifica per età dei mediatori del turnover lipidico da parte della metformina, abbiamo chiesto se la metformina influisce sulla deposizione lipidica sistemica in modo dipendente dall’età.
Eseguendo la colorazione lipidica di tutto il corpo Oil Red O in C. elegans abbiamo rilevato un calo significativo dei livelli di lipidi in animali giovani esposti a metformina, mentre non è stato possibile osservare tale declino negli animali anziani (Figura 6A, Figura S12A).
È importante sottolineare che una riduzione del contenuto lipidico di tutto il corpo da parte della metformina è coerente con il suo ruolo di mimetico della restrizione dietetica (DR) (Onken e Driscoll, 2010) che è stato recentemente ritenuto essenziale per l’estensione della longevità di questo farmaco (Pryor et al. , 2019). I nostri dati sugli animali anziani dimostrano che questo benefico effetto DR della metformina viene abolito durante l’invecchiamento.

Successivamente abbiamo eseguito l’analisi lipidomica mediante spettrometria di massa in nematodi giovani e vecchi esposti a metformina per caratterizzare in modo più dettagliato il fenotipo di turnover lipidico specifico per età indotto da questo farmaco. Confrontando l’abbondanza di lipidi in vermi giovani e vecchi non trattati abbiamo osservato un esaurimento significativo di fosfolipidi (PL), acidi grassi liberi (FFA), lisofosfolipidi e lisofosfolipidi ricchi di acidi grassi polinsaturi (PUFA) che si è verificato a causa dell’invecchiamento (Figura S13A-B e Tabella S4).
Da notare, queste osservazioni sono coerenti con un recente rapporto sulla lipidomica che caratterizza i cambiamenti dei lipidi basali nell’invecchiamento di C. elegans (Gao et al., 2017). È interessante notare che i livelli di trigliceridi (TAG) erano notevolmente aumentati negli animali anziani rispetto ai giovani vermi (Figura 6B, D e Tabella S4), in contrasto con altre classi di lipidi misurate e che ricordano l’accumulo aberrante di trigliceridi osservato nell’invecchiamento umano (Cree et al., 2004) e sull’insufficienza mitocondriale (Vankoningsloo et al., 2006). È importante sottolineare che l’accumulo di TAG durante l’invecchiamento è correlato all’elevato contenuto lipidico di tutto il corpo rilevato dalla colorazione Oil Red O nei vecchi nematodi non trattati (Figura 6A).
Successivamente abbiamo confrontato le risposte lipidiche alla metformina in animali giovani e anziani e abbiamo scoperto che nei giovani nematodi la metformina ha ridotto l’abbondanza di diverse sottoclassi lipidiche tra cui acidi grassi liberi e trigliceridi, coerente con il fenotipo DR-like (Figura 6B-D, Figura S13C-E , Tabella S4).
Sorprendentemente, i livelli di trigliceridi, compresi i TAG contenenti PUFA altamente insaturi, erano ulteriormente elevati nei vecchi animali esposti alla metformina (Figura 6B, D e Figura S13E), contrariamente alla risposta dei giovani nematodi e coerente con la perdita dell’effetto DR della metformina a vecchiaia.
I cambiamenti opposti nel contenuto di trigliceridi in animali giovani e anziani esposti a metformina erano anche coerenti con le dinamiche opposte delle proteine associate a goccioline lipidiche osservate in questi animali dalla proteomica (Figura 5G, Figura S9C, E): i trigliceridi sono i componenti chiave dei lipidi goccioline e il turnover delle goccioline lipidiche supportano il coinvolgimento dei TAG durante il digiuno e la DR (Lee et al., 2014).
Insieme all’aumento dei trigliceridi e dei trigliceridi ricchi di PUFA altamente insaturi (Figura S13E), la metformina ha innescato una riduzione dei lisofosfolipidi a catena lunga e ricchi di PUFA e degli acidi grassi liberi negli animali anziani (Figura S14A-C e Tabella S4), simile a riarrangiamenti lipidici aberranti osservati durante stress metabolico persistente (Markel et al., 1985; Nguyen et al., 2017; Steinhauser et al., 2018) e in linea con l’abbondanza differenziale di mediatori del turnover lipidico distinti rilevati in vecchi nematodi esposti a metformina da proteomica.
In sintesi, i vecchi animali non sono riusciti a sviluppare un fenotipo di turnover lipidico mimetico DR in risposta alla metformina, contrariamente ai giovani vermi, e piuttosto hanno mostrato una esacerbazione delle anomalie lipidiche associate all’invecchiamento preesistenti durante il trattamento con metformina.
Data l’importanza chiave recentemente riportata del turnover lipidico, simile alla DR, per l’estensione della vita da metformina (Pryor et al., 2019), l’inversione di questa risposta DR durante l’invecchiamento probabilmente contribuisce alla mancanza di benefici della metformina nella tarda età.
I cambiamenti lipidici innescati dalla metformina sono guidati da meccanismi distinti in giovane età e in età avanzata
Studi precedenti hanno implicato l’AMPK nei cambiamenti lipidici legati all’invecchiamento e alle alterazioni mitocondriali (Gao et al., 2017; Weir et al., 2017).
L’AMPK è stato anche collegato a una riduzione della sintesi di lipidi de-novo negli epatociti, muscoli e fegato esposti a metformina (Boudaba et al., 2018; Collier et al., 2006; Fullerton et al., 2013; Zang et al., 2004). Successivamente abbiamo chiesto se le trasformazioni lipidiche innescate dalla metformina in giovane età o in età avanzata fossero mediate da AMPK.
Animali selvatici e carenti di AMPK (AAK-2) sono stati trattati con metformina in giovane età e in età avanzata, seguita da colorazione lipidica di tutto il corpo Oil Red O. Abbiamo scoperto che i vecchi animali carenti di AMPK accumulavano lipidi aggiuntivi in risposta alla metformina mentre non si osservavano tali cambiamenti nei nematodi di tipo selvatico (Figura S11A, S12C), in linea con un ruolo importante di AMPK nel mitigare l’accumulo aberrante di lipidi durante lo stress metabolico della tarda età .
Ma curiosamente, i giovani nematodi carenti di AMPK hanno dimostrato una riduzione dei lipidi ancora più forte in risposta alla metformina rispetto ai controlli wild type (Figura 6E, S11C e S12B) indicando che AMPK non è l’istigatore principale della risposta alla metformina DR nei primi anni di vita, ma piuttosto svolge un ruolo inibitorio in questo processo.
Poiché il turnover lipidico specifico dell’età indotto dalla metformina influenzava i trigliceridi più forti di altre specie lipidiche, ci siamo poi chiesti se il percorso della lipolisi dei trigliceridi regolato dalla proteina chinasi A (PKA) in risposta al digiuno (Lee et al., 2014) è il principale driver dell’effetto della metformina nella prima infanzia sulle riserve lipidiche.
Eseguendo la colorazione lipidica di tutto il corpo Oil Red O nei nematodi di controllo e nei nematodi che ospitano RNAi abbattimento di kin-1 (l’ortologo di C. elegans di PKA) o atgl-1 (l’ortologo della lipasi trigliceridica adiposa, l’effettore chiave del PKA lipolysis pathway) abbiamo scoperto che l’inattivazione del pathway PKA effettivamente sopprimeva il turnover lipidico DR-simile indotto dalla metformina nei primi anni di vita (Figura 6F, S11B e S12D-E).
È interessante notare che in precedenza è stato dimostrato che AMPK modula direttamente l’attività di ATGL-1 verso un turnover più moderato delle riserve di trigliceridi e questa capacità inibitoria di AMPK è risultata essenziale per la lunga longevità delle larve di C. elegans dauer di lunga vita (Narbonne e Roy, 2009).
Nei nostri test abbiamo osservato un calo più marcato del contenuto lipidico in giovani animali con deficit di AMPK esposti a metformina rispetto ai controlli wild-type (Figura S11C), è quindi possibile che l’effetto modulatorio di AMPK sulla via della lipolisi PKA sia alla base del noto ruolo essenziale di AMPK nell’estensione della longevità da metformina (Onken e Driscoll, 2010), prevenendo l’esaurimento prematuro dei lipidi durante l’esposizione precoce alla metformina.
È importante sottolineare che i giovani animali esposti a metformina in presenza di HT115 E. coli (il veicolo RNAi), una condizione precedentemente trovata priva dell’estensione della vita di metformina simile ai mutanti AMPK (Cabreiro et al., 2013), hanno mostrato una perdita marcatamente aumentata di lipidi a 24 ore di trattamento farmacologico rispetto al controllo di E. coli OP50 e simili agli animali carenti di AMPK (Figura S11D e S12D-E, controllo del vettore vuoto); questa eccessiva perdita di lipidi è stata chiaramente prevenuta dall’inattivazione di ATGL-1, la lipasi trigliceridica chiave regolata da AMPK (Kim et al., 2016) (Figura S11D e S12D).
I nostri dati supportano quindi il modello di estensione della vita della metformina in cui la prevenzione della perdita di lipidi prematura e malsana è importante per una migliore longevità quanto l’induzione della risposta benefica al DR, precedentemente collegata al prolungamento della vita indotto dalla metformina (Onken e Driscoll, 2010; Pryor et al. ., 2019).
Mostriamo anche che AMPK ha ruoli specifici per età nel turnover lipidico attivato da metformina e che il percorso PKA e non AMPK istiga la risposta di utilizzo dei lipidi DR-simile in giovani animali trattati con metformina.
L’esaurimento dell’ATP e la tossicità della metformina nella tarda età sono alleviati dal co-trattamento in vivo con rapamicina
Poiché un appropriato turnover lipidico è essenziale per supportare i mitocondri con i metaboliti necessari per un’efficace sintesi di ATP, la nostra analisi molecolare delle vie chiave modulate dalla metformina ha ulteriormente supportato il ruolo primario dell’ATP esaurimento nel deterioramento cellulare innescato dalla metformina in tarda età.
Successivamente abbiamo deciso di testare se gli interventi in vivo accessibili che stabilizzano i livelli di ATP cellulare alleviano la tossicità della metformina in età avanzata. Un intervento noto che stabilizza il contenuto di ATP in condizioni di disfunzione mitocondriale e carenza di carbonio è l’inibitore TOR rapamicina (Thomsson et al., 2005; Zheng et al., 2016).
È interessante notare che l’inibizione del TOR è stata precedentemente ritenuta essenziale per supportare lo sviluppo dei nematodi e la sopravvivenza delle cellule dopo il trattamento con alte dosi tossiche di metformina (Wu et al., 2016). Studi precedenti hanno anche indicato che la co-esposizione alla rapamicina aumenta i benefici in termini di longevità del trattamento con metformina nella prima infanzia (Strong et al., 2016).
Successivamente abbiamo esposto fibroblasti presenescenti alla metformina in presenza o assenza di rapamicina e abbiamo osservato una chiara attenuazione della tossicità della metformina nelle cellule pretrattate con rapamicina (Figura 7A e S15A-B) accompagnata da esaurimento smorzato di ATP e ridotta perdita di potenziale di membrana mitocondriale in queste celle (Figura 7B-C e S15C-D).
Per verificare se la co-esposizione alla rapamicina protegge dalla tossicità della metformina nella tarda età anche in vivo, abbiamo pretrattato i nematodi con rapamicina dal giorno 8 dell’età adulta (AD8) seguito da un trattamento con metformina il giorno 10 dell’età adulta. Coerentemente con le nostre osservazioni sulle colture cellulari, la rapamicina il trattamento ha alleviato in modo significativo l’intolleranza alla metformina in età avanzata, come si è visto dal miglioramento della durata della vita mediana e massima negli animali co-esposti a rapamicina / metformina (Figura 7D).
Questi risultati in vivo confermano il ruolo decisivo dell’esaurimento dell’ATP nella tossicità della metformina in età avanzata e suggeriscono che gli interventi in grado di stabilizzare il contenuto di ATP cellulare, come la somministrazione di rapamicina, potrebbero essere utilizzati per la prevenzione dell’intolleranza alla metformina durante l’invecchiamento. L’inversione incompleta della tossicità della metformina nella tarda età da parte della rapamicina è in linea con la capacità limitata di questo farmaco di preservare i livelli di ATP cellulare.

link di riferimento: https://www.biorxiv.org/content/10.1101/863357v1.full



{kind=link}