










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Secondo un nuovo studio pubblicato su Neurology Clinical Practice, i supplementi che pretendono di migliorare la concentrazione mentale e la memoria possono contenere farmaci non approvati e in combinazioni e dosi potenzialmente pericolose.
I ricercatori hanno trovato cinque di questi farmaci non approvati negli Stati Uniti negli integratori esaminati.
Gli integratori sono talvolta chiamati “nootropici”, “farmaci intelligenti” o ” potenziatori cognitivi “.
“Gli integratori cognitivi da banco sono popolari perché promettono una mente più acuta, ma non sono così strettamente regolamentati come i farmaci”, ha detto l’autore dello studio Pieter A. Cohen, MD, della Harvard Medical School di Boston, Mass.
“L’uso di questi integratori pone rischi per la salute potenzialmente gravi.
Non solo abbiamo rilevato cinque farmaci non approvati in questi prodotti, ma abbiamo anche rilevato diversi farmaci che non erano menzionati sulle etichette e abbiamo trovato dosi di farmaci non approvati che erano fino a quattro volte superiori a quella che sarebbe considerata una dose tipica “.
Cohen ha detto che gli integratori potrebbero essere particolarmente rischiosi se usati in combinazione con farmaci da prescrizione o invece di consultare un medico.

rilevato; NA = Non applicabile; SD = deviazione standard
A differenza dei farmaci che devono essere dimostrati sicuri ed efficaci per l’uso previsto prima di essere commercializzati ai consumatori, la legge non richiede alla Food and Drug Administration (FDA) statunitense di approvare la sicurezza o l’efficacia degli integratori alimentari prima che raggiungano il consumatore.
La FDA interviene dopo che i prodotti raggiungono il mercato se sono etichettati in modo errato o contengono prodotti non approvati.
Per lo studio, i ricercatori hanno cercato nel National Institutes of Health Dietary Supplement Label Database e nel Natural Medicines Database per integratori cognitivi che elencavano farmaci simili al piracetam , un farmaco precedentemente trovato negli integratori ma non approvato negli Stati Uniti.
Stavano cercando analoghi del piracetam, farmaci con una struttura chimica simile ma leggermente diversa. A volte gli analoghi vengono introdotti nei supplementi per aggirare le leggi.
I ricercatori hanno identificato 10 integratori, otto che promettevano di migliorare la funzione mentale , uno che era stato commercializzato come “esplosivi per l’allenamento” e un altro che aveva le parole “sopravvivere, resistere, superare” sull’etichetta.
I ricercatori hanno esaminato il contenuto di ogni integratore utilizzando vari metodi e misurando le quantità di ciascun farmaco presente.
Nei 10 integratori esaminati, i ricercatori hanno rilevato cinque farmaci non approvati.
Due erano analoghi del piracetam chiamato omberacetam e aniracetam. Gli altri erano i farmaci non approvati vinpocetine , phenibut e picamilon .
La FDA ha emesso un avvertimento che la vinpocetina non dovrebbe essere consumata dalle donne in età fertile.
Sebbene tutti i rischi di questi farmaci non siano noti, gli effetti collaterali includono aumento e diminuzione della pressione sanguigna, agitazione, sedazione e ospedalizzazione.
Tutti e 10 gli integratori contenevano omberacetam , prescritto in Russia per lesioni cerebrali traumatiche e disturbi dell’umore.
Una tipica dose farmaceutica sarebbe di 10 milligrammi (mg). Le dosi in una porzione raccomandata di integratori erano fino a 40 mg, quattro volte maggiori rispetto ai dosaggi farmaceutici.
Alcuni integratori contenevano più di un farmaco non approvato. Un prodotto ha combinato quattro farmaci non approvati.
“Con fino a quattro farmaci non approvati nei singoli prodotti e in combinazioni mai testate sugli esseri umani, le persone che usano questi integratori per il potenziamento cognitivo potrebbero esporsi a rischi per la salute potenzialmente gravi”, ha detto Cohen.
“Gli effetti del consumo di combinazioni non testate di farmaci non approvati a dosaggi imprevedibili sono semplicemente sconosciuti e le persone che assumono questi integratori dovrebbero essere avvertite”.
I ricercatori hanno anche scoperto che per quei prodotti con quantità di farmaco fornite sulle etichette, la maggior parte delle quantità dichiarate era imprecisa.
“Il fatto che questi integratori siano elencati nei database ufficiali non significa che l’etichettatura sia accurata o che i livelli di dosaggio degli ingredienti in questi integratori siano sicuri”, ha detto Cohen.
“La legge statunitense non consente l’introduzione di prodotti farmaceutici non approvati negli integratori alimentari, ma la legge pone l’onere di eliminare quei prodotti sulla FDA. La FDA ha emesso una serie di avvertimenti alle aziende che vendono integratori con farmaci non approvati, tuttavia tali farmaci rimangono apertamente elencati nei database come ingredienti negli integratori.
Il nostro studio solleva anche preoccupazioni in merito alla qualità e alla legalità degli integratori elencati nei database degli integratori “.
Una limitazione dello studio era che non ha esaminato tutti i farmaci non approvati che sono commercializzati negli integratori cognitivi.
Fonte: Garvan Institute of Medical Research
Riferimenti
- US Food and Drug Administration. La FDA interviene contro 17 aziende per la vendita illegale di prodotti che affermano di curare la malattia di Alzheimer. 11 febbraio 2019. https: // www.fda.gov/news-events/press-announcements/fda-takes-action-against-17- companies- illegally -selling-products-claiming-treat-alzheimers-disease
- Cohen PA, Zakharevich I, Gerona R. Presenza di piracetam negli integratori alimentari di potenziamento cognitivo. JAMA Intern Med 2020; 180 (3): 458-459.
- Neznamov GG, Teleshova ES. Studi comparativi di noopept e piracetam nel trattamento di pazienti con disturbo cognitivo lieve nelle malattie cerebrali organiche di origine vascolare e traumatica. Neurosci Beh Physiol 2009; 39 (3): 311-321.
- Nakamura K. Aniracetam: il suo nuovo potenziale terapeutico nei disturbi disfunzionali cerebrali basato su recenti scoperte farmacologiche. CNS Drug Reviews 2002; 8 (1): 70-89.
- Avula B, Chittiboyina AG, Sagi S, et al. Identificazione e quantificazione di vinpocetine e picamilon negli integratori alimentari venduti negli Stati Uniti. Drug Test Anal 2016; 8 (3-4): 334-343. DOI: 10.1002 / dta.1853.
- Cohen PA. Vinpocetina: un farmaco non approvato venduto come integratore alimentare. Mayo Clinic Proc 2015; 90 (10): 1455.
- McCabe DJ, Bangh SA, Arens AM, Cole JB. Esposizioni Phenibut ed effetti clinici segnalati a un centro antiveleni regionale. Amer J Emerg Med 2019; 37 (11): 2066-2071.
- Cohen PA, Sharfstein J, Kamugisha A, Vanhee C.Analisi degli ingredienti degli integratori nel National Institutes of Health Supplement Database commercializzato come contenente una nuova alternativa agli steroidi anabolizzanti. JAMA Netw Open 2020; 3 (4): e202818.
- US Food and Drug Administration. Picamilon negli integratori alimentari. 29 novembre 2017. Disponibile su: https: // www.fda.gov/food/dietary-supplement-products-ingredienti / picamilon-dietary-supplementi
- US Food and Drug Administration. Phenibut negli integratori alimentari. 29 aprile 2019, disponibile su: https: // w ww .fda.gov / food / dietary-supplement-products- components / phenibut-dietary-supplementi
- US Food and Drug Administration. Dichiarazione sull’avvertimento per le donne in età fertile sui possibili rischi per la sicurezza degli integratori alimentari contenenti vinpocetina. 3 giugno 2019. Disponibile su: https: // w ww .fda.gov / news-events / press-annunciaments / statement-warning- women- childbearing -age-about-possible-safety- Countries -dietary-supplementi-contenenti
- Cohen PA, Bass S. Iniezione di sicurezza negli integratori – modernizzazione della legge sugli integratori alimentari. Nuovo Engl J Med 2019; 381 (25): 2387-2389.
- Cohen PA. La FDA e gli integratori adulterati – inosservanza del dovere. JAMA Netw Open. 2018; 1 (6): e183329.
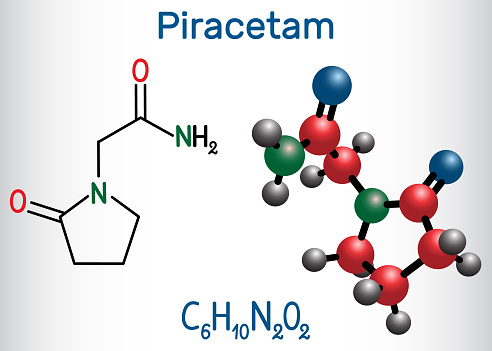
Piracetam
Il piracetam (2-osso-1-pirrolidineacetamide) (Figura 1) è un derivato ciclico dell’acido gamma-amminobutirrico (GABA), ottenuto dopo la perdita di una molecola di acqua seguita dalla formazione dell’anello (1).
È il primo rappresentante dei farmaci “nootropici” (2).
Il termine nootropico deriva da una parola greca che significa “agire sulla mente”.
Piracetam è stato sintetizzato da Giurgea nei laboratori UCB in Belgio. È in uso clinico dal 1972. Da allora altre società farmaceutiche si sono affrettate a sviluppare i propri nootropi (ad es. Vinpocetina, aniracetam, pramiracetam, oxiracetam).
Il piracetam può essere utilizzato con successo per trattare la demenza senile, le vertigini, l’anemia falciforme e numerosi altri problemi di salute come il morbo di Alzheimer o l’ictus (3-6).
Piracetam potrebbe aumentare la comprensione e l’accuratezza della lettura nei bambini dislessici (7).
Ha migliorato la vigilanza, la socializzazione e il QI nei pazienti psichiatrici anziani. Piracetam è stato anche usato per trattare l’alcolismo.
Nel 1991 Paula-Barbosa e colleghi hanno scoperto che l’alimentazione a lungo termine (12 mesi) di alcol per i ratti ha aumentato significativamente la formazione di lipofuscina (un pigmento di scarto correlato all’età) nelle cellule cerebrali.
Dare alte dosi di piracetam ai ratti nutriti con alcol ha ridotto i loro livelli di lipofuscina significativamente al di sotto dei livelli di controllo (8). Nel 1997 è stato dimostrato che il piracetam potrebbe ridurre la perdita neuronale a seguito del consumo cronico di alcol (9).
Piracetam migliora la cognizione in condizioni di ipossia e migliora anche la memoria e l’apprendimento (10). Quando il piracetam viene assunto con la colina, si verifica un effetto sinergico che provoca un maggiore miglioramento della memoria.
Le proprietà farmacologiche specifiche del piracetam sono state segnalate quasi 30 anni fa, ma il suo meccanismo d’azione era sconosciuto per molto tempo.
Piracetam è stato inizialmente testato in un modello di “nistagmo centrale” che era sensibile solo ai farmaci anticolinergici e antistaminici (2, 11).
L’uso corrente del piracetam nelle vertigini potrebbe essere correlato a questa proprietà.
La ricerca successiva, tuttavia, ha rivelato che il piracetam era privo di proprietà anticolinergiche o antistaminiche (5, 10, 12).
Sebbene il piracetam sia un derivato del neurotrasmettitore inibitorio GABA, il meccanismo della sua azione non è correlato a quello del GABA.
Il piracetam ha poca affinità per i recettori del glutammato, ma ha vari effetti sulla neurotrasmissione del glutammato.
Un sottotipo di recettore del glutammato è il recettore AMPA. Quantità micromolari (livelli che si ottengono tramite l’assunzione orale di piracetam) di piracetam aumentano l’efficacia dell’afflusso di calcio indotto da AMPA nelle cellule cerebrali.
Il piracetam aumenta anche la densità massima dei recettori AMPA nelle membrane sinaptiche dalla corteccia di ratto a causa del reclutamento di un sottoinsieme di recettori AMPA che normalmente non contribuiscono alla trasmissione sinaptica (12).
A livelli micromolari, il piracetam potenzia il rilascio di glutammato indotto dal potassio dai nervi dell’ippocampo di ratto (12).
Il piracetam è generalmente segnalato per avere effetti collaterali minimi o nulli.
È interessante notare, tuttavia, che gli effetti collaterali occasionalmente riportati dal piracetam di ansia, insonnia, agitazione, irritabilità e tremore sono identici ai sintomi di eccessiva neuroattività di acetilcolina / glutammato.
Nonostante questi effetti, il piracetam non è generalmente considerato un agonista o un inibitore significativo dell’azione sinaptica della maggior parte dei neurotrasmettitori.
I farmaci nootropici di tipo piracetam potrebbero esercitare il loro effetto su alcune specie di molecole presenti nella membrana plasmatica.
Sembrerebbe che agiscano come potenziatori di un’attività già presente, piuttosto che possedere una propria attività simile a un neurotrasmettitore (12). Pertanto, il piracetam non è soggetto agli effetti collaterali spesso gravi di farmaci che amplificano o inibiscono direttamente l’azione dei neurotrasmettitori, ad esempio inibitori MAO, inibitori selettivi della ricaptazione della serotonina, antidepressivi triciclici o anfetamine.
È stato scoperto che il piracetam facilita invece il trasferimento interemisferico, aumenta la resistenza cerebrale a stimoli nocivi come l’ipossia e migliora l’apprendimento e altre funzioni cognitive in condizioni normali (13, 14).
Tuttavia, il miglioramento di queste funzioni è molto più pronunciato quando la funzione cerebrale è compromessa da una varietà di stimoli nocivi (es. Ipossia, invecchiamento, lesioni cerebrali).
Gli esperimenti in vivo indicano che le proprietà di potenziamento cognitivo del piracetam sono solitamente più significative negli animali più anziani (15, 16). Suggerisce che il meccanismo d’azione dei farmaci nootropici sia associato ad alterazioni biochimiche nel cervello invecchiato.
Pertanto i nootropi potrebbero probabilmente ripristinare o contrastare questi cambiamenti biochimici. La ridotta fluidità delle membrane delle cellule cerebrali (probabilmente causata da concentrazioni più elevate di acidi grassi saturi) rappresenta un meccanismo associato ad alterazioni funzionali nel cervello in età avanzata (17) e potrebbe essere responsabile di deficit o disfunzioni dei meccanismi di trasduzione del segnale (18,19).
Piracetam nell’afasia
La maggior parte dei pazienti dopo l’ictus ischemico riacquista alcune delle funzioni perse. Il miglioramento della funzione sensomotoria è accompagnato da un aumento del flusso sanguigno nelle regioni cerebrali compromesse.
Mentre l’effetto della fisioterapia per il miglioramento dei deficit sensomotori è incontestato, l’efficacia della logopedia è ancora discutibile / poco chiara.
Se la riabilitazione possa essere potenziata dalla farmacoterapia adiuvante in pazienti con disturbi cerebrali è anche una questione di speculazione (20-22). Le prime sperimentazioni furono avviate negli anni Quaranta e riguardavano vari agenti in vari disturbi neurologici (23 – 25).

Poiché il piracetam migliora l’apprendimento e la memoria, Kessler et al. ha indagato in uno studio in doppio cieco, controllato con placebo, se il piracetam migliora il recupero del linguaggio nell’afasia post-ictus (26). Hanno scoperto che il piracetam migliora significativamente il flusso sanguigno attivato e facilita la riabilitazione dei pazienti afasici post ictus (26).
Il piracetam come adiuvante della logopedia migliora il recupero di varie funzioni del linguaggio e questo effetto è accompagnato da un aumento significativo dell’attivazione del flusso correlato al compito nelle aree eloquenti dell’emisfero sinistro (26).
Tuttavia, il meccanismo con cui piracetam migliora il recupero dall’afasia è ancora oggetto di speculazione. Poiché il tessuto infranto non può rigenerarsi, il recupero dall’afasia postroke deve coinvolgere regioni al di fuori dell’area morfologicamente danneggiata che probabilmente assumono le funzioni linguistiche perse nell’ictus acuto.

bloccanti GPIIb / IIIa.
Esistono alcune prove che piracetam agisca sulle piastrine come antagonista del trombossano A2 o come inibitore della sintetasi trombossano A2
insieme a una riduzione del livello plasmatico del fattore di von Willebrand (36). Il piracetam possiede anche un effetto reologico correlato alla sua
azione sulla deformabilità della membrana cellulare (37).
AA – Acido arachidonico; AC – Adenilil ciclasi; ADP – Adenosina difosfato; βTG – β-tromboglobulina; COX – cicloossigenasi; DAG
– Diacilglicerolo; GPIIb / IIIa – Glicoproteina IIb / IIIa; Gq, Gs – proteine G; 5-HT ñ Serotonina; IP3 – Inositolo-1,4,5-trifosfato; P2Y1, 12 –
Recettore ADP; PAF – Fattore di attivazione piastrinica; PF4 – Fattore piastrinico 4; PG G2, PG H2, PG I2, PG E2 – Prostaglandine G2, H2, I2, E2; PIP2
– Fosfatidilinositolo-4,5-difosfato; PKCa, PKCi – Protein chinasi C (rispettivamente attiva e inattiva); PLA2 – Fosfolipasi A2;
PLCβ, γ – Fosfolipasi Cβ, γ; TxA2 – Trombossano A2
Levetiracetam – derivato del piracetam con proprietà antiepilettiche
Levetiracetam è l’enantiomero S dell’α-etil-2-osso-1-pirrolidineacetamide (Figura 2).
Sebbene il piracetam possa essere utile nel mioclono e potenzi l’azione anticonvulsivante di vari farmaci antiepilettici (28,29), non era precedentemente usato di per sé nell’epilessia.
Levetiracetam, tuttavia, è stato approvato per il trattamento aggiuntivo dell’epilessia parziale, sia negli Stati Uniti che in Europa (27). Levetiracetam ha effetti antiepilettogeni e neuroprotettivi, con il potenziale di rallentare o arrestare la progressione della malattia (30).
Può giovare al mioclono nell’epilessia mioclonica progressiva (31). Sebbene il meccanismo d’azione del levetiracetam non sia completamente compreso, si suggerisce che una riduzione delle correnti di potassio nei neuroni possa contribuire ai suoi effetti antiepilettici (32).
Piracetam nella malattia di Alzheimer
Le membrane ippocampali dei pazienti con malattia di Alzheimer mostrano una ridotta fluidità che differisce dalle alterazioni della membrana specifiche dell’età. I dati clinici suggeriscono che il trattamento a lungo termine con piracetam sembra rallentare la progressione della malattia di Alzheimer, che si proponeva fosse spiegata dal ripristino della fluidità della membrana (19).
Piracetam in ictus
È stato riportato che il piracetam aumenta il flusso sanguigno cerebrale regionale compromesso nei pazienti con ictus acuto e, somministrato subito dopo l’esordio, migliora l’esito clinico (33, 34).
Potrebbe essere dovuto alla modifica delle proprietà reologiche del sangue circolante, modificando le risposte piastriniche (aggregazione e adesione) e all’effetto benefico sulla deformabilità dei globuli rossi, che porta a una riduzione attiva del rilascio di ADP da parte degli eritrociti danneggiati (35-37).
I dati sperimentali suggeriscono che l’efficacia del piracetam nella profilassi dell’ictus secondario non è buona come quella dell’acido acetilsalicilico (ASA) ma che il piracetam è meglio tollerato (38). Pertanto, il piracetam potrebbe essere un’alternativa per la profilassi dell’ictus secondario nei pazienti che non possono essere trattati con ASA o altri farmaci antipiastrinici.
Piracetam e piastrine
La modifica della funzione piastrinica, inclusa l’inibizione dell’aggregazione piastrinica da parte del piracetam, è nota da oltre 20 anni (35). Tuttavia, la modalità della sua azione è ancora dibattuta (36 – 39).
Piracetam normalizza le piastrine iperattive in vari disturbi tra cui ictus acuto (33, 34), attacchi ischemici cerebrali transitori, fenomeno di Raynaud e diabete mellito (36).
È stato suggerito che gli effetti inibitori delle piastrine siano dovuti a una ridotta reattività all’ADP o all’inibizione della sintesi del trombossano A2 (37, 40). È stato anche riportato che il piracetam ha un effetto diretto sulla parete vascolare, stimolando la produzione di prostaciclina nell’endotelio con una riduzione concomitante del rilascio del fattore di von Willebrand dai corpi di WeibelñPalade (37).
Le dosi necessarie per ottenere effetti reologici e / o antipiastrinici sono circa 2-4 volte superiori alle dosi necessarie per ottenere effetti nootropici (35). Tuttavia, anche a queste dosi elevate, il piracetam è ben tollerato e solo pochi effetti avversi sono stati registrati nei soggetti umani (5, 10).
Si ritiene che le proprietà reologiche del piracetam siano correlate ad alterazioni della membrana plasmatica come la struttura lipidica (37). Piracetam incorpora all’interno delle membrane a livello delle teste polari dei fosfolipidi. Questa interazione con i fosfolipidi di membrana ripristina la fluidità della membrana e potrebbe spiegare le proprietà farmacologiche del piracetam.
L’osservazione che il piracetam si lega ai gruppi della testa polare dei doppi strati della membrana e induce cambiamenti nella struttura della membrana può spiegare che questo farmaco funziona non solo nel cervello ma anche a livello delle cellule del sangue (19).
La sua attività è molto più pronunciata quando le membrane sono danneggiate (ad esempio con l’invecchiamento).
Poiché i cambiamenti a livello della membrana piastrinica sembrano essere coinvolti nella cascata di eventi che portano all’adesione e all’aggregazione piastrinica, questo effetto di modifica della membrana potrebbe essere il meccanismo d’azione principale del piracetam (Figura 3). Questo meccanismo differenzierebbe il piracetam da altri farmaci usati per inibire l’aggregazione piastrinica come ASA, ticlopidina o tirofiban. La ticlopidina, tuttavia, come farmaco lipofilo potrebbe anche alterare la fluidità della membrana piastrinica.
RIFERIMENTI
- Strubbe J.H., Cyprisiak E.: Rev. Indust. Chim. Belge 32, 112 (1967).
- Giurgea C.: Curr. Sviluppare. Psychopharmacol. 3, 221 (1976).
- Benesova O .: Drugs Aging 4, 285 (1994).
- Croisile B., Trillet M., Fondarai J., Laurent B., Mauguiare F., Billardon M .: Neurology 43, 301 (1993).
- Vernon MW, Sorkin EM: Drugs Aging 1, 17 (1991).
- Skondia V., Kabes J .: J. Int. Con. Ris. 13, 185 (1985).
- 7. DeBerdt W .: Life Sci. 55, 2057 (1994).
- Paula-Barbosa M .: Alcolismo Res. 15, 834 (1991).
- Cadete-Leite A., Brandao F., Andrade J., Ribeiro-da-Silva A., Paula-Barbosa M.: Alco- hol Alcohol. 32, 471 (1997).
- Noble S., Benfield P .: CNS Drugs 9, 497 (1998).
- Giurgea C .: Drug Dev. Ris. 2, 441 (1982).
- Gouliaev AH, Senning A .: Brain Res. Rev.19, 180 (1994).
- Nalini K., Karanth KS, Rao A., Aroor AR: Pharmacol. Biochem. 42, 859 (1992).
- Soerensen JB, Smith DF: J. Neural Transm. 6, 139 (1993).
- Roux S., Hubert I., Lenegre A., Milinkevitch D., Porsolt RD: Pharmacol. Biochem. Behav. 49, 683 (1994).
- Muller WE, Koch S., Scheuer K., Rostock A., Bartsch R .: Biochem. Pharmacol. 53: 135 (1997).
- Ghosh C., Dick RM, All SF: Neurochem. Int. 23, 479 (1993).
- Miyamoto A., Araiso T., Koyama T., Ohshika H.: J. Neurochem.55, 70 (1990).
- Muller WE, Eckert GP, Eckert A .: Pharma- copsychiatry 32, 2 (1999).
- Ferro J.M., Mariano G., Madureira S.: Cere- brovasc. Dis. 9, 6 (1999).
- 21. Small SL: Stroke 25, 1282 (1994).
- Wallesch CW, Muller U., Hermann M .: CNS Drugs 7, 203 (1997).
- Linn L .: Arch. Neurol. Psichiatria 58, 357 (1947).
- Bachman L., Morgan A .: Aphasiology 2, 225 (1988).
- Jacobs DH, Shuren J., Gold M., Adair JC, Bowers D., Williamson DJG, Heilman KM: Neurocase 2, 83 (1996).
- Kessler J., Thiel A., Karbe H., Heiss WD: Stroke 31, 2112 (2000).
- Shorvon SD, van Rijckevorsel K .: J. Neurol. Neurosurg. Psych. 72, 426 (2002).
- Chaudhry H .: Curr. Ther. Ris. 9, 355 (1992).
- Fedi M., Reutens D., Dubeau F., Andermann E., DíAgostino D., Andermann F .: Arch. Neurol. 58, 781 (2001).
- Ben-Menachem E .: Expert Opin. Pharmacother. 4, 2079 (2003).
- Crest C., Dupont S., Leguern E., Adam C., Baulac M .: Neurology 62, 640 (2004).
- Madeja M., Margineanu DG, Gorji A., Siep E., Boerrigter P., Klitgaard H., Speckmann EJ: Neuropharmacology 45, 661 (2003).
- Orgogozo JM: Stroke 32, 1449 (2001).
- Ferguson JJ: Circulation 94, 1195 (1996).
- Bick, RL: Lancet 2, 752 (1979).
- Evers S., Grotemeyer KH: Pharmacopsychia- try 32, 44 (1999).
- Moriau M., Crasborn L., Lavenne-Pardonge E., von Frenckell R., Col-Debeys C.: Pharmaceutical research 43, 110 (1993).
- Grotemeyer KH, Evers S., Fischer M., Hus- stedt IW: J. Neurol. Sci. 181, 65 (2000).
- Peuvot J., Schanck A., Deleers M., Brasseur R .: Biochem. Pharmacol. 50, 1129 (1995).
- Bick RL, Thromb. Haemost. 46, 67 (1981).
Vinpocetine
La vinpocetina (14-etossicarbonil- (3a, 16a-etil) -14,15-eburnammina) è un derivato sintetico dell’alcaloide della vinca vincamina che è un alcaloide estratto dalla pianta della pervinca, Vinca minor (Fig.1). La vinpocetina può attraversare la barriera emato-encefalica ed entrare nel cervello dopo somministrazione orale o endovenosa (Gulyas et al., 2002a, Gulyas et al., 2002b).
La struttura chimica, la farmacocinetica, il metabolismo e la distribuzione sono stati precedentemente esaminati in dettaglio (Bonoczk et al., 2000). Vinpocetine, nome commerciale come Cavinton, è stato originariamente sviluppato e commercializzato in Ungheria intorno al 1978. Vinpocetine è stato clinicamente utilizzato in molti paesi asiatici ed europei per la prevenzione e il trattamento di ictus, demenza senile e disturbi della memoria.
Inoltre, numerose marche di pillole o prodotti per la memoria contenenti vinpocetina sono attualmente disponibili in tutto il mondo anche come integratori alimentari ( https://naturalmedicines.therapeuticsresearch.com ).
Non sono stati segnalati effetti collaterali e tossicità significativi per la vinpocetina a dosi terapeutiche e generalmente si ritiene che sia sicura per l’uso a lungo termine.
La vinpocetina ha quindi attirato una notevole attenzione da parte della comunità accademica e scientifica, nonché delle industrie farmaceutiche, per caratterizzare le sue nuove funzioni terapeutiche, il meccanismo di azione e gli obiettivi farmacologici.

Strutture chimiche di vincamina e vinpocetina
Bersagli farmacologici della vinpocetina La
vinpocetina ha un numero di bersagli cellulari differenti (Bonoczk et al., 2000, Patyar et al., 2011) (Tabella 1). La fosfodiesterasi 1 ciclica del nucleotide (PDE1) è tra i primi target farmacologici riportati per la vinpocetina (Ahn et al., 1989, Chiu et al., 1988, Hagiwara et al., 1984, Souness et al., 1989).
Le PDE sono una superfamiglia di enzimi che catalizzano la degradazione di cAMP e / o cGMP, che sono raggruppati in undici ampie famiglie, PDE1 – PDE11, in base alle loro proprietà cinetiche distinte, meccanismi regolatori e sensibilità agli inibitori selettivi (Bender e Beavo, 2006) .
I membri della famiglia PDE1 sono codificati da tre geni distinti, PDE1A, PDE1B e PDE1C con più varianti di splicing N-terminale e / o C-terminale (Chan e Yan, 2011).
L’attività catalitica della PDE1 può essere stimolata dal calcio in presenza di calmodulina, motivo per cui PDE1 viene anche chiamata PDE stimolata da Ca2 + / calmodulina. L’attivazione Ca2 + -dipendente degli isoenzimi PDE1 gioca un ruolo critico nel crosstalk tra Ca2 + e la segnalazione ciclica dei nucleotidi (Yan et al., 2003).
I singoli isoenzimi PDE1 differiscono per l’affinità del substrato, la sensibilità al Ca2 + e la distribuzione tessuto / cellula. In vitro, PDE1A e PDE1B mostrano affinità di substrato molto più elevate per cGMP rispetto a cAMP, mentre PDE1C è ugualmente sensibile sia per cGMP che per cAMP (Chan e Yan, 2011).
La vinpocetina ha affinità inibitorie distinte per diverse isoforme PDE1. Ad esempio, la vinpocetina inibisce la PDE1A o la PDE1B a IC50 – 8-20 μM, mentre la PDE1C a IC50 ≈40-50 μM (Loughney et al., 1996, Yan et al., 1996, Yu et al., 1997).
Pertanto, la vinpocetina ha un’affinità maggiore per PDE1A / 1B rispetto a PDE1C. Inoltre, la vinpocetina può anche agire come un bloccante per i canali Na + dipendenti dalla tensione. Ad esempio, studi precedenti attraverso approcci con patch clamp hanno dimostrato che la vinpocetina bloccava i canali Na + dipendenti dalla tensione a valori IC50 di 10-50 μM (Molnar e Erdo, 1995, Sitges et al., 2005, Sitges e Nekrassov, 1999, Zhou et al. , 2003).
Più recentemente, è stato segnalato che la vinpocetina è un inibitore della chinasi IκB (IKK), con un valore IC50 intorno a 17 μM (Jeon et al., 2010). IKK svolge un ruolo fondamentale nella mediazione delle risposte infiammatorie cellulari.
In risposta a stimoli infiammatori esterni, viene attivato un insieme di complessi IKK. Il complesso IKK attivato fosforila IκBα, portando alla sua ubiquitinazione e degradazione. IκB è un inibitore di NF-κB che è un fattore trascrizionale chiave responsabile dell’espressione di una varietà di mediatori proinfiammatori, comprese citochine, chemochine e molecole di adesione.
NF-κB viene liberato a causa della degradazione di IκB e quindi entra nel nucleo per attivare la trascrizione delle molecole infiammatorie (Rothwarf e Karin, 1999). Pertanto, la fosforilazione di IκBα mediata da IKK è il punto centrale nella regolazione della risposta infiammatoria dipendente da NF-κB. Pertanto, la vinpocetina, inibendo l’attività IKK, agisce come un nuovo potente agente antinfiammatorio (Jeon et al., 2010, Medina, 2010).
Tabella 1
Obiettivi molecolari multipli della vinpocetina
| Obiettivi | Valori IC 50 | Espressione di tessuti / cellule |
|---|---|---|
| Isoforme PDE1 | PDE1A / 1B: 8-20 μM | Cervello: PDE1A, 1B e 1C; SMC contrattili: |
| PDE1C: 40-50 μM | PDE1A; SMC proliferanti: PDE1A e 1C; Cuore: PDE1A% 1C; Macrofagi: PDE1B | |
| Canale Na + | 10-50 μM | Cervello, cuore, vasi |
| IKK | ≈ 17 μM | La maggior parte dei tipi di cellule |
Vinpocetina e malattie neurologiche La
vinpocetina è stata inizialmente sviluppata per il trattamento di malattie neurologiche associate a disturbi cerebrovascolari come ictus e demenza che sono spesso causati da ischemia o altri deficit cognitivi.
Numerosi studi hanno riportato gli effetti protettivi della vinpocetina dopo un danno ischemico del cervello nei roditori (Jincai et al., 2014, Rischke e Krieglstein, 1991, Sauer et al., 1988) e nell’uomo (Bonoczk et al., 2002, Szilagyi et al., 2005, Szobor e Klein, 1976, Vas e Gulyas, 2005, Vas et al., 2002, Zhang et al., 2016).
Inoltre, la vinpocetina sembra essere utile anche per i disturbi neuronali degenerativi come il morbo di Parkinson (PD) (Medina, 2011, Sharma e Deshmukh, 2015), la malattia di Huntington (HD) (Gupta e Sharma, 2014) e la malattia di Alzheimer (AD ) (Heckman et al., 2015, Medina, 2011).
Nel cervello, la vinpocetina migliora il flusso sanguigno cerebrale agendo come un vasodilatatore cerebrale (Bonoczk et al., 2000, Bonoczk et al., 2002, Patyar et al., 2011, Szilagyi et al., Vas et al., 2002, Zhang e Yang, 2015); e migliora il metabolismo cerebrale aumentando l’assorbimento di ossigeno e glucosio e stimolando la produzione di ATP neuronale (Bonoczk et al., 2000, Bonoczk et al., 2002, Patyar et al., 2011, Szilagyi et al., 2005, Zhang e Yang, 2015) .
In un certo numero di cellule neuronali o terminali nervosi, la vinpocetina ha anche dimostrato di funzionare come antiossidante (Deshmukh et al., 2009, Herrera-Mundo e Sitges, 2013, Horvath et al., 2002, Pereira et al., 2000, Santos et al., 2000, Solanki et al., 2011) e prevengono l’aumento neurotossico di calcio e sodio (Sitges et al., 2005, Sitges e Nekrassov, 1999, Tretter e Adam-Vizi, 1998).
È quindi evidente che molteplici azioni meccanicistiche della vinpocetina attraverso diversi bersagli molecolari contribuiscono agli effetti neuroprotettivi della vinpocetina.
Inoltre, la vinpocetina provoca anche effetti protettivi in altre condizioni correlate all’ischemia, come la retina (Nivison-Smith et al., 2014, Nivison-Smith et al., 2017, Nivison-Smith et al., 2015), il fegato (Abdel Salam et al., 2007, Zaki e Abdelsalam, 2013), reni (Fattori et al., 2017) e pelle (Xiao-Xiao et al., 2013).
Vinpocetina e infiammazione
Recentemente, la vinpocetina si è dimostrata un potente agente antinfiammatorio in una varietà di cellule coltivate in vitro come le cellule endoteliali vascolari (EC) (Jeon et al., 2010), le cellule muscolari lisce vascolari (SMC) (Jeon et al., 2010), monociti / macrofagi (Jeon et al., 2010), neutrofili (Ruiz-Miyazawa et al., 2015), cellule epiteliali (Jeon et al., 2010, Liu et al., 2014), cervello cellule microgliali (Zhao et al., 2011) e cellule dendritiche (Feng et al., 2017).
Attraverso l’inibizione diretta dell’attività IKK, la vinpocetina attenua la fosforilazione di IκB mediata da IKK e aumenta la stabilità di IκB, che porta al legame di IκB con NF-κB e la successiva soppressione dell’espressione della molecola infiammatoria dipendente da NF-κB.
L’effetto della vinpocetina sull’attività trascrizionale antagonizzante dipendente da NF-κB è improbabile mediato prendendo di mira i canali PDE1 o Na + come inibitore selettivo per PDE1 IC86340 e tetrodotossina bloccante del canale Na + non hanno avuto effetto sull’attività trascrizionale di NF-κB (Jeon et al., 2010) .
Gli effetti antinfiammatori della vinpocetina sono stati rivelati anche in vari modelli animali in vivo. In un modello di lesione da ischemia-riperfusione cerebrale di ratto, è stato riscontrato che i livelli di NF-KB e TNFα erano associati a cambiamenti nell’edema cerebrale e nel volume dell’infarto.
La vinpocetina ha inibito l’espressione di NF-KB e TNFα e ha ridotto la risposta infiammatoria dopo ischemia-riperfusione cerebrale (Wang et al., 2014a).
Ancora più importante, l’effetto antinfiammatorio della vinpocetina è stato recentemente riportato in uno studio multicentrico che ha coinvolto 60 pazienti con occlusione della circolazione cerebrale anteriore e insorgenza di ictus (Zhang et al., 2017). I pazienti trattati con vinpocetina non solo hanno avuto un migliore recupero della funzione neurologica e migliori risultati clinici, ma hanno anche ridotto l’attivazione del segnale NF-κB e l’espressione del mediatore pro-infiammatorio.
Inoltre, la vinpocetina è efficace anche in altri modelli di malattie infiammatorie animali, come il dolore infiammatorio indotto da lipopolisaccaridi (LPS) (Ruiz-Miyazawa et al., 2015), infiammazione polmonare indotta da LPS (Jeon et al., 2010), otite di topo media (Lee et al., 2015) e danno renale acuto (Fattori et al., 2017).
Vinpocetina e malattie vascolari
L’effetto vasorilassante della vinpocetina è stato dimostrato anche in vasi periferici di specie diverse, probabilmente mediato dall’inibizione della PDE1 che idrolizza preferenzialmente cGMP con elevate affinità (Ahn et al., 1989, Chiu et al., 1988, Giachini et al., 2011, Hagiwara et al., 1984, Souness et al., 1989).
È stato precedentemente dimostrato che la PDE1A era sovraregolata in un modello di ratto di tolleranza alla nitroglicerina (NTG) e la vinpocetina ripristinava parzialmente la sensibilità del sistema vascolare tollerante alla successiva esposizione a NTG (Kim et al., 2001).
L’attivazione di PDE1A può ridurre i livelli di cGMP. Pertanto, l’induzione di PDE1A in vasi tolleranti ai nitrati può essere un meccanismo mediante il quale la vasodilatazione mediata da NO / cGMP viene desensibilizzata e la vasocostrizione mediata da Ca2 + è supersensibilizzata. L’inibizione dell’attività PDE1A potrebbe essere efficace per limitare la tolleranza ai nitrati.
È stato segnalato che altri inibitori selettivi della PDE1 come IC86340 (Miller et al., 2009) e Lu AF41228 / Lu AF58027 (Laursen et al., 2017) causano vasodilatazione e / o abbassamento della pressione sanguigna nei roditori. Il ruolo della PDE1A nella regolazione della pressione sanguigna è stato più specificamente confermato dalla recente scoperta che i topi con assenza di attività PDE1A avevano una pressione sanguigna aortica più bassa (Wang et al., 2017).
Studi genetici sull’uomo hanno rivelato l’associazione dei polimorfismi a singolo nucleotide PDE1A (SNP) con la pressione diastolica (Bautista Nino et al., 2015, Yan, 2015). Inoltre, la vinpocetina ha anche aumentato i livelli di cGMP e la risposta vasodilatatrice polmonare dopo l’inalazione di NO negli agnelli con ipertensione polmonare acuta, probabilmente attraverso l’inibizione della PDE1 (Evgenov et al., 2006).
L’analisi dell’associazione genome-wide ha identificato il locus PDE1A associato all’ipertensione arteriosa polmonare idiopatica in una popolazione giapponese (Kimura et al., 2017).
Oltre alla vasodilatazione, studi recenti hanno anche rivelato le nuove funzioni della vinpocetina in altre malattie vascolari croniche associate al rimodellamento strutturale vascolare. Ad esempio, un recente studio di Cai et al. ha indicato che la vinpocetina ha attenuato in modo significativo l’ispessimento della parete del vaso e la formazione di neointimal nelle arterie carotidi di topo dopo una lesione da legatura e il rimodellamento spontaneo marcatamente soppresso degli espianti di vene safene umane in un modello di coltura ex vivo (Cai et al., 2012).
Uno studio simile ha anche riportato che la vinpocetina ha attenuato l’iperplasia neointimale dell’arteria carotide indotta da lesioni da palloncino nei ratti diabetici (Wang et al., 2014b). Nelle SMC coltivate, la vinpocetina ha inibito la crescita delle SMC attraverso la produzione di specie reattive dell’ossigeno antagonizzante (ROS) (Cai et al., 2012, Wang et al., 2014b).
Gli effetti inibitori della vinpocetina sulla crescita di SMC e sul rimodellamento vascolare sembrano essere in linea con le conseguenze della soppressione specifica di PDE1A o PDE1C. L’abbattimento genetico dell’espressione di PDE1A attraverso l’interferenza specifica dell’RNA ha attenuato la crescita delle SMC vascolari e promosso la morte delle SMC (Nagel et al., 2006). L’esaurimento della PDE1C ha inoltre soppresso in modo significativo la crescita e la migrazione di SMC in vitro (Cai et al., 2015).
Nei topi knockout PDE1C globali che hanno tassi di crescita normali, schemi di alimentazione, comportamenti normali di allattamento e accoppiamento, la formazione neointimale indotta da lesioni della legatura carotidea è stata soppressa di ≈75% rispetto ai topi di controllo wild-type (Cai et al., 2015).
Tuttavia, non è noto se l’effetto antiossidante della vinpocetina nelle SMC sia mediato dall’inibizione della PDE1. Studi genetici sull’uomo hanno definito associazioni significative di PDE1A SNP con spessore intima-media carotideo (Bautista Nino et al., 2015, Yan, 2015).
È interessante notare che la vinpocetina ha anche ridotto la trombosi vascolare dopo lesioni vascolari nei topi (Cai et al., 2012), il che è coerente con l’effetto precedentemente riportato della vinpocetina sull’aggregazione piastrinica antagonista (Akopov e Gabrielian, 1992). Inoltre, è stato dimostrato che la vinpocetina attenua l’aterosclerosi indotta dalla dieta ad alto contenuto di grassi nei topi con deficit di ApoE (Cai et al., 2013, Zhuang et al., 2013), nonché previene l’aterosclerosi e la calcificazione nei modelli di aterosclerosi del coniglio (Yasui et al. al., 1989, Yasui et al., 1990).
La vinpocetina ha dimostrato di sopprimere l’accumulo di lipidi nei macrofagi (Cai et al., 2013) e la differenziazione osteoblastica delle SMC (Ma et al., 2016), gli eventi chiave nell’aterogenesi vascolare umana. Pertanto, la vinpocetina può rappresentare un promettente farmaco o integratore per la terapia clinica dell’aterosclerosi e della calcificazione vascolare.
Vinpocetina e malattie cardiache Un
recente studio di Wu et al. ha studiato i ruoli della vinpocetina nell’ipertrofia cardiaca, nella fibrosi e nel rimodellamento cardiaco patologico in vitro e in vivo (Wu et al., 2017).
È stato dimostrato che la vinpocetina sopprime in modo significativo l’ipertrofia miocardica del topo e la fibrosi cardiaca in vivo indotte dall’infusione cronica di angiotensina (Ang II). Studi meccanicistici hanno inoltre dimostrato che la vinpocetina ha attenuato direttamente la crescita ipertrofica dei cardiomiociti adulti di topo e l’attivazione dei fibroblasti cardiaci in vitro, probabilmente in modo PDE1-dipendente.
La scoperta di questo studio è coerente con i precedenti rapporti sulla funzione PDE1A e PDE1C nel rimodellamento cardiaco con approcci genetici specifici. Ad esempio, è stato dimostrato che PDE1A e 1C sono sovraregolati nei cuori ipertrofici degli animali indotti da isoproterenolo (ISO) – o ang II-infusione e / o cuori in cedimento indotto da sovraccarico di pressione cronico o infarto miocardico, così come nel fallimento umano cuori (Knight et al., 2016, Miller et al., 2011, Miller et al., 2009).
Il knockdown specifico della PDE1A riduce l’ipertrofia dei cardiomiociti (Miller et al., 2009), l’attivazione dei fibroblasti e la produzione di matrice extracellulare (Miller et al., 2011).
Allo stesso modo, l’esaurimento genetico della PDE1C ha attenuato l’ipertrofia, la morte e la fibrosi dei cardiomiociti sia in vitro che in vivo (Knight et al., 2016). Inoltre, la vinpocetina ha anche provocato effetti inibitori sulle correnti di sodio nei cardiomiociti, che possono essere protettivi per il cuore (Ver Donck et al., 1993, Wei et al., 1997).
Tuttavia, il ruolo della vinpocetina nella morte dei cardiomiociti rimane sconosciuto. La morte massiva dei cardiomiociti è associata a ischemia cardiaca / danno da riperfusione. Dati gli effetti anti-apoptotici della vinpocetina in vari altri tessuti (Lakics et al., 1995a, Onishchenko et al., 2008, Sonmez et al., 2017) e tipi di cellule (Bora et al., 2016, Chen et al., 2007, Erdo et al., 1990, Gabryel et al., 2002, Lakics et al., 1995b, Miyamoto et al., 1989), gli effetti protettivi della vinpocetina sulla morte cardiomiocitica e sul danno cardiaco I / R rimangono da indagare .
Uno studio recente ha rilevato che la PDE1A è altamente espressa nelle cellule nodali senoatriali cardiache del coniglio e regola la funzione del pacemaker. Pertanto, la funzione della vinpocetina nella regolazione della frequenza cardiaca merita di essere ulteriormente studiata.
Conclusione e prospettiva
Evidentemente, la vinpocetina è un agente multi-azione con una varietà di bersagli farmacologici.
Le sue multi-azioni, tra cui vasodilatazione, antiossidazione, antinfiammatoria, antitrombosi e antimodellante, possono agire insieme per suscitare effetti terapeutici sinergici, fornendo così benefici significativi a quelle malattie multifattoriali cerebrovascolari e cardiovascolari.
Inoltre, la vinpocetina è efficace per un’ampia gamma di condizioni patologiche. La spiegazione potrebbe essere che molte malattie condividono patologie comuni che possono essere migliorate dalla vinpocetina, come antagonizzare l’infiammazione in una varietà di tipi di cellule, proteggere diverse cellule dalla morte durante le lesioni da ischemia e stimolare la vasodilatazione per aumentare il flusso sanguigno in diversi tessuti.
Va notato che le recenti scoperte volte a esplorare nuove funzioni della vinpocetina dipendono in gran parte da modelli animali. Sono necessari studi clinici sull’uomo per convalidare l’efficacia della vinpocetina nel prevenire il rimodellamento patologico vascolare e cardiaco, nonché in varie malattie infiammatorie.
I meccanismi molecolari alla base di alcuni degli effetti farmacologici della vinpocetina, inclusi il potenziamento metabolico, l’antiossidazione e l’antitrombosi, rimangono ancora poco chiari e devono essere ulteriormente studiati. Lo sviluppo di successo di animali geneticamente modificati, inclusi topi knockout genici o topi transgenici di isoforme PDE1, IKK, e i canali del sodio faciliterebbero la delucidazione di un’azione meccanicistica più precisa della vinpocetina.

Vinpocetina è un agente multi-azione con una serie di bersagli farmacologici
Vinpocetina ha tre principali bersagli molecolari tra cui PDE1, canale Na + voltaggio-dipendente e IKK. È stato dimostrato che la PDE1 regola la vasocostrizione, il rimodellamento della struttura vascolare e cardiaca e la neurotrasmissione. Il canale Na + è coinvolto nella tossicità cellulare e nella morte cellulare. IKK svolge un ruolo fondamentale nella mediazione della risposta infiammatoria cellulare.
link di riferimento: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5766389/
Phenibut può essere descritto come un derivato fenilico dell’acido gamma-amminobutirrico (acido γ- amminobutirrico o GABA). Il nome del composto “pheni-but” si basa sulla sua origine di sintesi e sulla sua struttura, che fu riportata per la prima volta da Perekalin et al negli anni ’60 (Lapin, 2001).
L’aggiunta di un anello fenilico all’acido butirrico consente al composto di attraversare la barriera emato-encefalica. Phenibut agisce come un agonista GABA, principalmente sui recettori GABA b , stimolando il rilascio di dopamina. Al contrario, il GABA stesso non attraversa la barriera emato-encefalica, limitando il suo utilizzo come farmaco terapeutico praticabile per trattare l’ansia. Tuttavia, il GABA si trova naturalmente anche nel sistema nervoso umano.
In questo momento, GABA è un farmaco controllato B1 ai sensi del Misuse of Drugs Act 1975, poiché è una sostanza da cui è possibile derivare il gamma-idrossibutirrato o GHB (comunemente noto come Fantasy). GHB è un farmaco controllato B1 ai sensi del Misuse of Drugs Act 1975.

Phenibut è classificato come GABApentinoid. I GABApentinoidi sono una classe di farmaci che si lega e blocca i canali del calcio voltaggio-dipendenti contenenti la subunità α2δ. Questa classe include Baclofen che è indicato per la spasticità dei muscoli scheletrici (p.es., Lioresal), Pregabalin che è indicato per il dolore neuropatico e per il controllo delle crisi epilettiche, (p.es., Lyrica) e Gabapentin un anticonvulsivante, analgesico e ansiolitico per il trattamento dell’ansia disturbi (p. es., Neurontin). Baclofen, Pregabalin e Gabapentin sono tutti attualmente programmati come farmaci da prescrizione nella Tabella 1 dei Regolamenti sui medicinali del 1984.

Gli usi più comuni esposti per il phenibut sono come nootropico (un farmaco usato per migliorare la memoria o altre funzioni cognitive), per ridurre l’ansia, per migliorare il sonno, aumentare il desiderio sessuale e aumentare la produzione dell’ormone della crescita umano. Questi ultimi due sembrano essere accademicamente infondati.
Nome comune internazionale (o nome approvato dal Regno Unito o nome adottato dagli Stati Uniti) del medicinale
Nessuna.
Nomi chimici
- Acido 4-ammino-3-fenilbutirrico
- Acido 4-ammino-3-fenilbutanoico [nome IUPAC]
- acido beta- (amminometil) benzenepropanoico
- acido beta- (amminometil) idrocinnamico
- beta-fenil-gamma-amminobutirrato
- acido beta-fenil-gamma-amminobutirrico
- beta-fenil-GABA
- β-fenil-γ-amminobutirrato
- Acido β-fenil-γ-amminobutirrico
Altri nomi
- Phenibut / Fenigam
- Phenigam / Phenigama / Phenygam
- Phenylgamma
- PhGaba / Pgaba
- Noofen
Formula chimica Peso molecolare
C10H13NO2 179,2 gmol -1
Struttura chimica

Stato di classificazione in altri paesi (in particolare Australia, Regno Unito, Stati Uniti, Canada)
Australia – L’Advisory Committee on Medicines Scheduling (luglio 2017) ha raccomandato che il phenibut fosse classificato come sostanza proibita Schedule 9, da usare solo per scopi di ricerca. Il rapporto del TGA sulla raccomandazione del Comitato è incluso come allegato a questa presentazione.
USA – Non programmato. Phenibut è disponibile come integratore alimentare in quanto soddisfa i criteri del Dietary Supplement Health and Education Act 1994 (DSHEA) come derivato sintetico dell’amminoacido. Di solito è promosso con indicazioni sul mantenimento della salute (p. Es., Aiuta a mantenere la calma) invece di affermazioni sullo stato di malattia o sulla condizione (p. Es., Riduce l’ansia) (Cutter 2016, Owen et al, 2016).
Regno Unito – Non programmato. Nel 2014, l’MHRA ha sequestrato una vasta gamma di potenziatori cognitivi, incluso il phenibut, sulla base del fatto che si trattava di prodotti medicinali senza licenza forniti per la vendita (MHRA 2014).
Non è chiaro se il phenibut sia catturato ai sensi dello Psychoactive Substances Act 2016 del Regno Unito. Alcune delle attività segnalate della sostanza rientrano in categorie di effetti regolamentati dalla presente legge, come i cambiamenti di vigilanza, umore, empatia e sonnolenza (effetti che si applicherebbero a molti nootropi). Il possesso di potenziatori cognitivi non è illegale, sebbene l’azione dell’MHRA indichi che la vendita senza licenza è vietata (ad esempio, se il fenibut è stato effettivamente determinato come una sostanza potenziatore cognitivo).
Canada – Non programmato. La Direzione per i prodotti sanitari naturali e non soggetti a prescrizione medica del Canada ha osservato che ciò non soddisfa i loro criteri di considerazione come sostanza sanitaria naturale in quanto non è una sostanza presente in natura (NHPID 2017). È disponibile come sostanza non programmata, di solito presso rivenditori in linea, negozi di alimenti naturali e forum di body building.
Europa – Non programmato. Non è regolamentato dall’Agenzia europea per i medicinali. Tuttavia, alcune sostanze nootropiche come il piracetam sono disponibili solo su prescrizione in alcuni paesi dell’Unione europea e sono disponibili gratuitamente in altri.
Russia – Phenibut è un farmaco con prescrizione medica usato per una varietà di condizioni tra cui ansia, insonnia, disturbi da stress post-traumatico, depressione, balbuzie, tic, disturbi da deficit di attenzione e disturbi vestibolari. Si dice che i cosmonauti russi abbiano ricevuto la sostanza per alleviare la tensione, l’ansia e la paura (Buckley 2006).
Lettonia – come per la Russia.
Non ci sono informazioni sul sistema di segnalazione degli eventi avversi (FAERS) della FDA su Phenibut.
Non ci sono informazioni sul database delle reazioni avverse di phenibut CARM.
Nel database Vigilyze dell’OMS, ci sono stati 19 ICSR (Individual Case Safety Report) segnalati su phenibut dal 2012, con un aumento in numero dal 2016. La maggior parte delle segnalazioni riguardava persone di età compresa tra 18 e 44 anni provenienti dalle Americhe, e il la maggior parte degli eventi era associata a disturbi generali, del sistema nervoso o psichiatrici.
Motivi per richiedere la classificazione
Phenibut agisce come un agonista completo del recettore GABA b , responsabile dei suoi effetti sedativi. A dosi più elevate, il phenibut agisce anche come un GABA un agonista (Shulgina 1986).
Gli utenti di vari siti Web e forum in linea (ad esempio Reddit.com, Social Anxiety Support.com, Brain Pro Tips.com, Bluelight.com, Erowid.org, Corpina.com) hanno riportato effetti positivi:
- calma estrema
- maggiore socialità
- maggiore senso di “benessere”
- lieve euforia
- aumento variabile della vigilanza
- cognizione migliorata e ritenzione della memoria
Gli effetti negativi riportati da utenti e ricercatori (Samokhvalov et al 2013; Sankary et al 2017; Joshi et al 2017) sono:
- postumi della sbornia
- dipendenza
- mal di testa
- depressione, ansia
- sedazione, in particolare da dose eccessiva o uso eccessivo
- letargia
- agitazione
- delirio
- confusione
- raramente, convulsioni tonico-cloniche
La maggior parte degli articoli e delle discussioni in linea si riferiscono alla tolleranza (effetto ridotto) con un uso a lungo termine e sono necessarie dosi crescenti per ottenere l’effetto precedentemente sperimentato. Ci sono anche effetti di astinenza, che possono includere agitazione, rabbia, ansia, depressione, vertigini, allucinazioni, insonnia, irritabilità, nausea, tremori.
Si prevede che l’astinenza dal phenibut si presenti come l’astinenza da baclofen (Alvis e Sobey, 2017) e quella di altri agonisti del GABA b e gestita con benzodiazepine e cure di supporto (Maryland Poison Center 2017; O’Connell 2017; American Addiction Centers 2017, Mental Health Daily 2016).
L’entità dell’uso del fenibut in Nuova Zelanda è sconosciuta. Tuttavia, è disponibile per la vendita presso i rivenditori in linea come Trademe.co.nz, supplement.co.nz, iherbs.co.nz, tripme.co.nz, biovea.com, evitamins.com, nootsupply.co. nz.
La maggior parte del phenibut disponibile in commercio sembra essere nella forma del sale cloridrato, phenibut HCl. L’ingestione di fenibut cloridrato a stomaco vuoto o con caffeina (che stimola la secrezione gastrica) aumenta la velocità di assorbimento. Phenibut è anche disponibile in commercio come amminoacido libero, che è più lento a dissolversi ed è leggermente amaro al gusto. A differenza del sale cloridrato, l’amminoacido libero può essere assorbito per via nasale, sublinguale e rettale (Psychonautwiki 2017).
Le dosi iniziali variano in modo significativo nei vari forum degli utenti Web (ad esempio, Reddit.com, LiftMode.com, Bluelight.com, Corpina.com, Nootrohacker.com). 200-500 mg sono stati suggeriti per l’effetto nootropico. Per altri effetti, incluso come ansiolitico, antidepressivo, coadiuvante del sonno per l’insonnia, droghe ricreative, sono state raccomandate dosi di 250-750 mg fino a tre volte al giorno. In alcuni atleti è stato segnalato un uso fino a 3000 mg al giorno (Tomen, 2017).
Phenibut potrebbe anche potenziare o migliorare gli effetti di tranquillanti, narcotici e farmaci neurolettici (Psychonautwiki 2017).
discussione e conclusioni
Le affermazioni nootropiche (e altre indicazioni terapeutiche) sono associate al phenibut.
Phenibut ha un effetto psicoattivo e potrebbe essere considerato una sostanza psicoattiva ai sensi dello Psychoactive Substances Act 2013 se venduto allo scopo di indurre un effetto psicoattivo e la sua importazione potrebbe essere interrotta in base a tale legge. Tuttavia, il vero scopo dietro un’importazione è spesso difficile da stabilire quando le spedizioni vengono fermate alla frontiera dalla dogana. Poiché attualmente è una sostanza non programmata, l’importatore può affermare che è stata importata per uso terapeutico personale.
Dalla letteratura, sembrano esserci alcuni rischi associati al suo uso, più comunemente mal di testa, letargia, dipendenza e sintomi di astinenza tra cui agitazione, irritabilità, rabbia. In casi estremi, sono state segnalate convulsioni.
Phenibut non soddisfa il rischio moderato di soglia di danno necessario per essere controllato ai sensi del Misuse of Drugs Act 1975. Tuttavia, il suo uso e la disponibilità dovrebbero essere controllati a causa del rischio di effetti avversi.
Il comitato di classificazione dei Medicinali ha discusso le sostanze migliorando nootrope e cognitive racetam e le strutture al 53 racetam simil- rd e 54 th incontri e avvisati che essere classificati come medicinali soggetti a prescrizione. Pertanto, la classificazione del phenibut come medicinale soggetto a prescrizione sarebbe coerente con l’approccio del Comitato alle sostanze nootropiche, la schedulazione della classe di sostanze GABApentinoid e gli usi ei rischi associati alla sostanza.
Riferimenti
Alvis BD, Sobey CM. (2017). Astinenza dal baclofene orale con conseguente debolezza progressiva e sedazione che richiedono il ricovero in terapia intensiva. Neuroospedalista. 7 (1): 39-
40. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5167087/
Centri americani per le dipendenze. (2017). Phenibut ritiro, riduzione e disintossicazione. Accesso 30 ottobre 2017.
https://americanaddictioncenters.org/withdrawal-timelines-treatments/phenibut/
Buckley JC. (2006). Fisiologia spaziale. Capitolo 2, pagina 44. Oxford University Press, New
York. https://books.google.com.au/books?id=Jn_i6KbutXYC&pg=PA44&lpg=PA44&d q = phenibut + spazio & source = bl & ots = HUOIOj01T2 & sig = ObESzW8x8jSeYbZXWVzmxz 2VQpw & hl = it & sa = X & ved = 0ahUKEwjMh6-255bXAhVLHZQKHWh9DRoQ6AEIVDAH # v = onepage & q = phenibut% 20spacephenib ut & f = falso
Cutter A. (2016). Phenibut non è né provato né sicuro come farmaco miracoloso prosociale. 25 novembre 2016. HealthButSmart.com. https://sciencebasedmedicine.org/phenibut-is-neither-proven-nor-safe-as-a-prosocial- wonder-drug /
Corpina (2015). Il buono e il cattivo lato di Phenibut. Accesso 30 ottobre 2017. https://corpina.com/positive-negative-side-effects-phenibut/
Joshi YB, amico SF, Jimenez B, Steiger LR. (2017). Intossicazione dissociativa e astinenza prolungata associata a phenibut: A Case Report. Journal of Clinical Psychopharmacology 37: 478-
80 https://www.ncbi.nlm.nih.gov/labs/articles/28614159/
Lapin I. (2001). Storia dello sviluppo di farmaci. Revisioni dei farmaci sul sistema nervoso centrale 7 (4): 471-481. Neva Press, Branford, Connecticut. http://onlinelibrary.wiley.com/doi/10.1111/j.1527- 3458.2001.tb00211.x / pdf
Liftmode 2017. L’ultima guida al dosaggio di phenibut e come prenderla. Accesso 30 ottobre 2017. https://liftmode.com/blog/phenibut-dosage-guide/
Maryland Poison Center. (2017). Phenibut – Wonder Drug o Unsafe Supplement? ToxTibits, agosto 2017, Maryland Poison Center, Università del Maryland School of Pharmacy. Accesso 30 ottobre
2017. http://www.mdpoison.com/media/SOP/mdpoisoncom/ToxTidbits/2017/August%202017%20ToxTidbits.pdf
Salute mentale quotidiana. (2016). Sintomi di astinenza da Phenibut: elenco delle possibilità. Accesso al 30 ottobre 2017. http://mentalhealthdaily.com/2015/12/21/phenibut- ritiro-sintomi-elenco-di-possibilità /
MHRA, (2014). Comunicato stampa: Il cane da guardia dei farmaci registra il sequestro record di farmaci intelligenti sperimentali. MHRA, 24 ottobre
2014. https://www.gov.uk/government/news/medicines-watchdog-makes-record- seizure-of-experimental-smart-drugs
NHPID (2017). Database degli ingredienti dei prodotti naturali per la salute. Phenibut. Accesso 30 ottobre 2017.
http://webprod.hc-sc.gc.ca/nhpid-bdipsn/ingredReq.do?id=11307&lang=eng
O’Connell CW, Schneir AB, Hwang JQ, Cantrell FL. (2014). Phenibut: attenzione dell’acquirente. Il blog dell’American Journal of Medicine. Accesso 30 ottobre
2017. http://amjmed.org/phenibut-buyer-beware/
Owen DR, Archer JR, Dargan PI. (2016). Phenibut (acido 4-ammino-3-fenil-butirrico): disponibilità, prevalenza d’uso, effetti desiderati e tossicità acuta. Revisione di droghe e alcol. 35 (5): 591-6. https://www.ncbi.nlm.nih.gov/pubmed/26693960
Psychonaut Wiki 2017. Phenibut. Accesso 30 ottobre 2017. https://psychonautwiki.org/wiki/Phenibut
Samokhvalov AV, Paton-Gay CL, Balchand K, Rhem K. (2013). Dipendenza da fenibut. Caso clinico BMJ
2013. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3604470/
Sankary S, Canino S, Jackson J. (2017). Overdose di fenibut. American Journal of Emergency Medicine 35: 516e1-516e2. http://www.ajemjournal.com/article/S0735- 6757 (16) 30574-5 / abstract
Shulgina GI. (1986). Sui meccanismi neurotrasmettitori di rinforzo e inibizione interna. La rivista pavloviana di scienze biologiche 21 (4): 129-40. (PubMed.gov / NCBI). https://www.ncbi.nlm.nih.gov/pubmed/2431377
Tomen D. (2017). Phenibut. Sito web Nootropicsexpert. Accesso al 30 ottobre 2017. http://nootropicsexpert.com/phenibut/ Database Vigilyze (2018). Phenibut. Accesso ai dati. 25 gennaio 2018. https://vigilyze.who-umc.org/#/



{kind=link}