










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A new paper in Nature Communications outlines how an international research team has identified potential ways forward to rapidly design improved and more potent compounds in the fight against COVID-19.
The work is the result of a massive fragment screening effort to develop an antiviral targeting the SARS-CoV-2 main protease.
The project was led by Martin Walsh, Deputy Life Sciences Director at Diamond Light Source; Frank von Delft, Professor of Structural Chemical Biology at the University of Oxford and Principal Beamline Scientist of I04-1/XChem at Diamond; and Nir London, Assistant Professor at the Weizmann Institute Israel.
The team combined mass spectrometry with the XChem facility at Diamond, the UK’s national synchrotron, to rapidly identify new lead compounds for drug development to treat COVID-19.
For this study, called Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease, the team probed an essential enzyme of SARS-CoV-2 with over 1,250 unique small compound, termed fragments, and identified 74 high-value fragment hits which can be used to develop new inhibitors for this essential viral protein.
The paper details the data along with proposed design routes for progressing towards improved, more potent, compounds.
“COVID-19, caused by SARS-CoV-2, lacks effective therapeutics. Additionally, no antiviral drugs or vaccines were developed against the closely related coronavirus, SARS-CoV-1 or MERS-CoV, despite previous zoonotic outbreaks.
To identify starting points for such therapeutics, we performed a large-scale screen of electrophile and non-covalent fragments through a combined mass spectrometry and X-ray approach against the SARS-CoV-2 main protease, one of two cysteine viral proteases essential for viral replication.
Our crystallographic screen identified 74 hits that span the entire active site, as well as three hits at the dimer interface. These structures reveal routes to rapidly develop more potent inhibitors and offer unprecedented structural and reactivity information for on-going structure-based drug design against SARS-CoV-2 main protease,” explains Martin Walsh who is addition to his role at Diamond is also a Medical Research Council (MRC) funded Research Group Leader at the Research Complex at Harwell (RCaH).

Structural biology, which can play a key role in drug development, was also rapidly deployed after the 2002 SARS-CoV-1 outbreak, with earlier work by the Hilgenfeld group on the main protease of coronarviruses leading to crystal structures of SARS-CoV-1 protease and inhibitor complexes.
Other studies have taken the popular approach of a high-throughput screens (HTS) using very large compound libraries, followed by structural studies to elucidate the binding mode.
“Despite these efforts, drugs remain elusive that directly target SARS-CoV-2 (rather than disease symptoms) and are verified by clinical trials.
In retrospect, this is perhaps unsurprising for the main protease inhibitors, as both peptidomimetic and covalent inhibition carry risks as strategies for drug development; in general, the simpler the molecule the lower the risk. We, therefore, applied a different approach to this protease, using fragment screening by high-throughput structural biology,” adds Martin Walsh.
Fragment methods have become a staple of modern drug discovery, using small collections (100 s or 1000 s) of small compounds (<300 Da) that bind promiscuously and thus sample a far larger chemical space than is achieved by HTS.
The challenge is that the very weak binding of fragment hits requires highly sensitive biophysical detection, careful confirmation of binding and specialised medicinal chemistry expertise take the hits and develop them into fully potent drug candidates.
However, the real promise of fragments with the right expertise and equipment, they can quickly and efficiently be converted into valid drug candidates with a much simpler route to clinical impact.
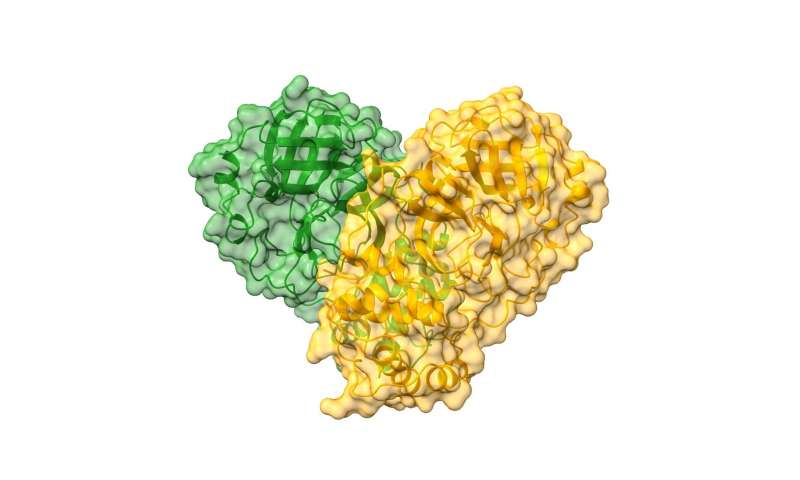
- Structure of SARS-CoV-2 Main protease: Cartoon representation of the CoVID-19 dimer with a semi-transparent surface in green and orange delineating each monomer. Credit: Diamond Light Source
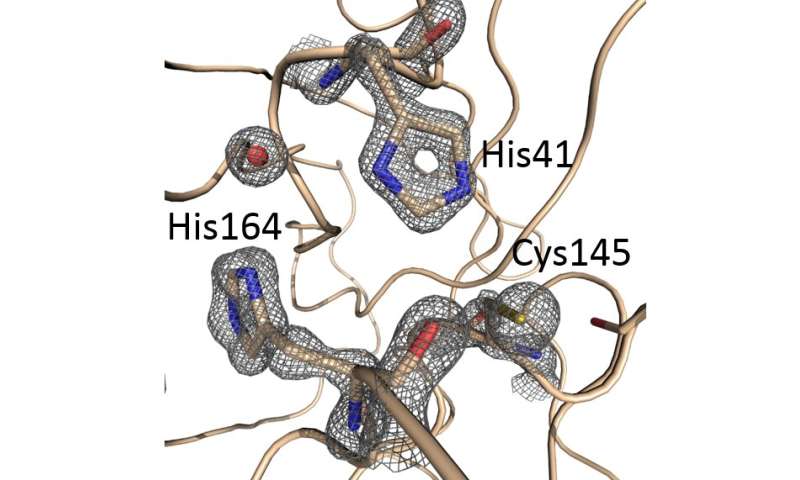
- Structure of SARS-CoV-2 Main protease: Representative electron density (2Fo-Fc map contoured at the 2.5 level) from the 1.39 Å structure centered at the active site of the enzyme. Credit: Diamond Light Source
Rapid advances in technology and automation at synchrotron radiation sources has made screening directly in crystal structures routinely possible at facilities like the XChem platform at Diamond Light Source.
The team took the highly unusual route of releasing all the experimental data as soon as it was generated; the announcement on social media triggered a large international collaboration that harnessed the combined knowledge of scientists worldwide through a novel crowdsourcing initiative that they called COVID Moonshot.
“Performing the experiment and achieving the high data quality in a few weeks, as lockdown started, was a tour de force, and a credit to our highly talented scientists.
Even more remarkable was the response of the international community to the data release: it mobilized a vast pool of expertise, technologies and philanthropy, which evolved into a unique and rigorous drug discovery effort that aims to develop rapidly an entirely novel, easily synthesized, oral antiviral with good safety and pre-clinical properties.
Working fully in the open, data are released near real-time, so the outcome will be available to any drugs manufacturer world-wide.
The world’s focus has been on vaccines and repurposing of existing drugs, but the Moonshot is one of a small number of projects attempting novel small molecule therapeutics,” says Frank von Delft.
SARS-CoV-2 is a large enveloped, positive-sense, single stranded RNA Betacoronavirus. The viral RNA encodes two open reading frames that, through ribosome frame-shifting, generates two polyproteins pp1a and pp1ab. These polyproteins produce most of the proteins of the replicase-transcriptase complex.
The polyproteins are processed by two viral cysteine proteases: a Papain-like protease (PLpro) which cleaves three sites, releasing non-structural proteins nsp1-3 and a 3C-like protease, also referred to as the main protease (Mpro), that cleaves at 11 sites to release non-structural proteins (nsp4-16).
These non-structural proteins form the replicase complex responsible for replication and transcription of the viral genome and have led to Mpro and PLPro being the primary targets for antiviral drug development.
Host and viral proteases involved in viral life-cycle
As for common viral infections, the crucial event for the viral life cycle is the entry of genetic material inside the host cell for replication and release of new virions. During its life-cycle, SARS-CoV-2 is internalized in the host cell where the viral RNA is translated, exploiting the host cell machinery and giving rise to virus-encoded proteins of different open reading frames (ORF)s.
The ORF1, which encompasses about 75% of the viral genome, is translated into two viral replicase polyproteins (i.e., pp1a and pp1ab) (Fig. 1 ).
Sixteen mature non-structural proteins (nsp) arise from further processing of these two pps, which are autocatalytically processed by two proteases (also auto-processed), namely
- (a) the papain-like protease (PLpro), which cleaves the first two non-structural proteins (nsp1 and nsp2) at the N-terminal region of the polyprotein, and
- (b) the main protease (Mpro, also known as a chymotrypsin-like cysteine protease, 3CLpro), which recognizes cleavage sites at the C-terminus and brings to the production of about 11 individual mature non-structural proteins [5], [3], [4].
The remaining ORFs encode accessory and structural proteins, like spike surface glycoprotein (S), small envelope protein (E), matrix protein (M), nucleocapsid protein (N) (see Fig. 1).

SARS-CoV-2 polyproteins encoded by ORF1a and ORF1ab. Schematic representation of the open reading frames 1a and 1ab, which encode for polyproteins pp1a and pp1ab. Proteins composing each polyprotein are shown: (ns) indicates non-structural proteins; RNA dependent RNA polymerase and Helicase are indicated by (RdRp) and (Hel), respectively. Proteolytic sites cleaved by PLpro and Mpro are reported in yellow and green arrows, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Since proteolytic enzymes are the major actors of the various events described in this review and although knowledge about their role is continuously expanding, it may be worth recalling that they can be roughly classified into seven broad groups (from the type of aminoacid involved as proton donor for the activation of the peptide bond to be cleaved), namely
- (a) serine protease,
- (b) cysteine protease,
- (c) threonine protease,
- (d) aspartic protease,
- (e) glutamic protease,
- (f) metalloproteases (usually employing Zn++), and
- (g) asparagine peptide lyases [6], [7].
Within each group, a further differentiation can be applied according to whether the peptide bond cleaved by the specific enzyme corresponds to a terminal residue (i.e., exoprotease) or else to one of aminoacids within the sequence (i.e., endoprotease).
Endoproteases targeted for the development of anti-viral strategies
The activity of several endoproteases ensures viral infection, involving host and viral proteases which belong to the classes of serine- and cysteine-proteases, respectively.
Both proteases of the host cell (which are supposed to assist the virus during the intracellular and extracellular phases of its cycle) and those of the virus act in a concerted fashion to regulate and coordinate specific steps of the viral propagation, such as
- (i) the entry and the replication of the virus,
- (ii) the maturation of the polyprotein,
- (iii) the assembly of the secreted virions for further diffusion [5] (Fig. 2 ).

Diagram of the involvement of host and viral proteases in SARS-CoV-2 life cycle. Activation of coronavirus spike proteins by host cell proteases occurs at different stages in the viral life cycle and in different cell localizations. The ACE2-dependent infectious entry at the cell membrane is triggered through the S protein cleavage by host proteases: furin (1) and/or TMPRSS2 (2). Intracellular activation of S protein is mediated by cathepsin in lysosomes (3) and/or by Furin in trans-Golgi network (TGN) (4). After the receptor recognition, the viral genome is released into the cytoplasm of the host cell (5), RNA attaches directly to the host ribosome for translation of two polyproteins (not shown). Polyprotein (pp) maturation into mature fragments is catalysed by viral Cys proteases (Mpro and PL pro) (6). RNA is translated into DNA and inside the nucleus (N) replication amplifies the number of virus genome copies (7). The viral genome produces pps, which help to take command over host ribosomes for their own translation process; protein biosynthesis starts at the endoplasmic reticulum (ER) and follows the constitutive secretory pathway along Golgi compartments (8). The virion assembly occurs (9) and the newly packed viral particles can egress (10).
The spike glycoproteins are responsible for the crown-like appearance of Coronavirus particles (Fig. 3 A), playing a crucial role for the entry of the viral genome inside the host cell (Fig. 2).
The first critical step is the binding of the homotrimeric S protein with its specific cellular receptor, which triggers a cascade of proteolytic events leading to the fusion of cell and viral membranes. Similarity in structure and sequence with SARS-CoV and in vitro binding measurements indicate that SARS-CoV-2 S protein shows an improved binding for the receptor of angiotensin converting enzyme2 (ACE2), identifying it as the main host receptor [8].
The S protein is synthesized as an uncleaved precursor which includes two functionally distinct domains (i.e., S1 and S2 domains) that are responsible for receptor binding and triggering of the fusion event, respectively (Fig. 3B).

(A): Schematic representation of coronavirus particle. Spike proteins are highly glycosylated type I transmembrane protein, which assemble into trimers on the virion surface to form the distinctive “corona” (crown-like) appearance. (B): Domain organization and cleavage sites of the coronavirus Spike monomer (S). The ectodomain of all CoV spike proteins share the same organization in two domains, that is a N-terminal domain, named S1 and responsible for receptor binding, and a C-terminal S2 domain responsible for fusion. The domain organization of the S monomer consists of a signal peptide (SP), the N-terminal domain (NTD), the receptor-binding domain (RBD), the fusion peptide (FP), the internal fusion peptide (IFP), the heptad repeat 1/2 (HR1/2), and the transmembrane domain (TM).The region between the two domain is termed S1/S2 site. (C): Sequence of S1/S2 cleavage site of S protein from SARS-CoV-2. The four amino acid insertion (SPRRs), unique to SARS-CoV-2, is marked in yellow, the conserved S1/S2 cleavage site is marked in grey. (D): Comparative sequences of S protein cleavage sites. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The inactive CoV S protein acquires both cellular receptor binding and fusion function upon cleavage events at different sites, which can be carried out by multiple proteases at multiple sites in different cell compartments [10], [9] (see Fig. 2). Importantly, depending on CoV strain and cell type, CoV S protein is activated at a specific cell localization by one or several host proteases, including furin, trypsin, cathepsin L, transmembrane protease serine protease2 (TMPRSS2), TMPRSS4, or human airway trypsin-like protease (HAT) [11] (see Fig. 2). Exploiting redundant pathways to activate surface glycoproteins, the activating cleavage is mediated by multiple host membrane proteases via two distinct pathways, namely either (i) the late endosomal pathway, using cathepsins, and/or (ii) the cell-surface or early endosome pathway, using transmembrane serine proteases (e.g., TMPRSS2 and pro-protein convertase furin) (Fig. 2).
It has been suggested that the surface route is preferred under natural conditions, while repeated passages in cultured cells in vitro appears to exert a selective pressure in favour of virions bearing a greater capacity to invade the target cell via late endosomes [12], [9], [13]. Thus, to activate the fusion machinery of the viral S protein the cooperation in space and in time of multiple membrane proteases is demanded; the actually involved pool depends on both the virus strain and the specific host cell type expression profile of proteases (thus changing for each cell type).
Among host proteases, involved in the viral infection, furin is the one most widely present, being constitutively expressed in a variety of cell types. It cycles from the trans Golgi network (TGN)/endosomal compartments and cell surface, and it is known to accumulate in the TGN (where it is supposed to fulfil its proteolytic activity) [14](Fig. 2). Nevertheless, recently it has been also detected linked to the membrane of oral and airway epithelial cells [16], [15].
Unlike close relatives, SARS-CoV-2 can promptly infect a broad spectrum of human cell types, spanning from lung cells to endothelial, conjunctival and gut cells, with the respiratory district being the main target, displaying the peculiar ability to infect even the upper respiratory tract. The efficient spreading of virus relies on the protease arsenal of host cells which mediate the propagation of viral infection. The expression profile of furin and ACE2 in human cells could explain why SARS-CoV-2 is so efficient in spreading virus particles, since they are present throughout the body in endothelial cells with particularly increased levels in cells lying in alveoli and small intestine[17]. Moreover, SARS-CoV-2 S protein possesses a peculiar insertion of four amino acids (i.e., Ser-Pro-Arg -Arg-Ala-Arg689↓, see [18] and Fig. 3C), which has been identified as an additional cleavage site for the specificity of furin activity, strengthening the idea that this enzyme plays a dominant relevance in SARS-CoV-2 viral infection [13], [19], [20], [21].
Therefore, furin may play either (a) a role in the first entry of the virus, thanks to its topological location at the outer membrane (which would allow the formation of the ternary complex with ACE2, i.e., furin:SARS-CoV-2S:ACE2), and/or (b) during the transport of virions along the secretory pathway, further facilitating the virus diffusion (Fig. 2). This co-expression has been detected in airway epithelia, cardiac tissues and enteric canals [16], envisaging the possibility that in these districts the role of furin in favouring the virus cell entry is relevant, providing a cellular and molecular basis for the comprehension of the major clinical effects of COVID-19 in the tissues where these cell types are located.
A key discovery in understanding the mechanism of SARS-CoV-2 infection concerns the role of the androgen-responsive transmembrane serine protease 2 (TMPRSS2), that is expressed by specific epithelial tissues (including those of the respiratory and digestive tracts), facilitating the SARS-CoV-2 entry in the human airways by cleaving the viral spike (S) protein [22], [23], [19] (Fig. 2).
Beside host proteolytic enzymes, two viral proteases, namely Mpro and PLpro (involved in the maturation of viral polyprotein) are also recognized as important drug target(s). In particular, Mpro has been found to play a prominent role in the viral gene expression and replication, thus becoming an attractive target for anti-CoV-2 drugs. Notably, its quaternary structure renders Mpro ideal for rational drug design strategies against SARS-CoV-2, as there is a correlation between homodimer formation and the enzyme catalytic activity. Each protomer contains an antiparallel β-barrel structure, which has a folding scaffold similar to other viral chymotrypsin-like proteases. However, unlike chymotrypsin, the active site of SARS-CoV-2 Mpro contains a catalytic cysteinyl residue instead of a serine residue.
It must be stressed that although the endoprotease classes show a variety of catalytic sites (see above) and distinct protein folding, functional similarity can be found across evolutionary distant species (from viruses to humans) [6], thus representing a caveat in the development of effective COVID-19 therapeutic strategies.
Further, structural and evolutionary analyses indicate that SARS-CoV-2 Mpro is a highly conserved viral protein, which recognizes the sequence Leu-Met-Phe-Gln↓Ser-Gly-Ala while no human proteases share the same specificity [24]. This unique feature makes Mpro an even more attractive target for a broad inhibition of multiple stages in the viral life cycle (such as viral formation, progression of the viral infection and reproduction of virions).
Overall, two very attractive processes (which indeed represent important targets for designing anti-viral drugs), will be discussed here: (a) the proteolytic activation of the S protein (by furin and TMPRRS2), impairing the entry of viral genetic material inside the host cell, and (b) the activity of viral proteases (in particular Mpro), impairing the formation of mature viral proteins, which are required for the progression of the viral infection and replication of viruses.
Activation of spike (S) protein by host proteases
Multiple cleavages of coronavirus S protein are the primary determinants of the viral tropism, since this protein is responsible for receptor binding and, once cleaved, it drives the fusion of the viral envelope with the cell membrane.
Structure-function of S protein
This large glycoprotein (approx. 180 kDa) is present on the viral surface as a prominent trimer, each monomer ectodomain being composed of S1 and S2 domains (Fig. 3B). Although the sequence comparison of the S protein between SARS-CoV and SARS-CoV-2 indicates only 76% identity (large variations exist at the N-terminus), it has been assumed that their folding is similar [25].
The approx. 1200 amino acid sequence of the spike glycoprotein consists of a large ectodomain, a single pass transmembrane anchor, and a short C-terminal intracellular tail [26], showing several conserved domains and different motifs, namely (a) a signal peptide (SP) (which commits the protein to the constitutive secretory pathway), (b) the N-terminal domain (NTD), (c) the receptor-binding domain (RBD), (d) the fusion peptide (FP), (e) the internal fusion peptide (IFP), (f) the heptad repeat 1/2 (HR1/2), (g) the transmembrane domain (TM), and (h) the endo-domain in the cytosol [9], [26] (Fig. 3B).
The globular N-terminal S1 subunit mediates the infection of receptor-expressing host cells; it contains a receptor-binding domain (RBD) (Fig. 3B), made by 193 amino acids fragment, which is responsible for recognizing and binding the cell surface receptor (e.g., ACE2, see below).
On the other hand, the S2 domain mediates viral–membrane fusion through the exposure of a highly conserved fusion peptide, important for the entry of the viral genome inside the host. Multiple cleavage events activate the viral S protein with the involvement of several host proteases; the cleavage at the S1/S2 sites splits S into S1 and the S2 bioactive fragments, while another critical cleavage event, conserved in all coronaviruses, occurs at the so called S2′ site [27] (see Fig. 3B).
The cleavage at the S1/S2 sites, also termed priming, can be mediated by ACE2 and furin (and/or other bio-membrane anchored proprotein convertases, namely PoCo5B and PoCo7) [19]. Thus, in SARS-CoV-2 S the cleavage S1/S2 site 1 for furin (i.e., Pro Arg Arg Ala Arg689 ↓Ser Val) is conserved with respect to other CoVs (Fig. 3D). On the other hand, like SARS-CoV, SARS-CoV-2 spike protein displays an additional putative cleavage recognition motif at the S1/S2 site 2 (Ile Ala Tyr ↓ Thr Met Ser) resulting from the four aminoacidic insertion (Ser Pro Arg Arg) (see Fig. 3C and D); this suggests that the SARS-CoV-2 might operate a peculiar mechanism to promote its entry into host cells [13], [28].
Moreover, both SARS-CoV-2 and MERS-CoV display Pro685 at the S1/S2 junction. The turn created by Pro685 is important for the glycations of flanking residues; notably, the O-linked glycans Ser673, Thr678 and Ser686 are unique to SARS-Cov-2 [29]. This feature might envisage an O-glycation somehow modulating the cleavability of the spike protein (Fig. 3C).
The C-terminal of the S2 domain, known as the biomembrane-anchored stalk domain, anchors the S protein to the lipid transmembrane and it is involved in the viral entry [30]. It contains the fusion peptide (FP), followed by an internal fusion peptide (IFP) and two heptan domains (HR)s preceding the transmembrane domain (TM) (see Fig. 3B).
Noteworthy, like SARS-CoV, the SARS-CoV-2 S2 domain contains a proteolytic site S2′, found immediately upstream of the fusion peptide [31], which displays the canonical furin-like cleavage site (i.e., Lys-Arg815↓Ser-Phe) and whose cleavage is critical for the fusion process (see Fig. 3C and 3D) [13].
In addition to the canonical furin S2′ cleavage site the SARS-CoV-2 S protein shows an additional potential furin-like cleavage site (referred as polybasic insert), absent in CoV of the same clade, which in turn broadens the spectrum of proteases that can be exploited by the virus [19], [13], [29] (see Fig. 3D). As a matter of fact, trypsin-like proteases (TMPRSS2) and cathepsin L have also been shown to be involved in SARS-CoV-2 infection [20], [11]. The formation of the S2 fragment and the further activation by the cleavage at the S2′ site is an important determinant of the transmissibility and pathogenicity of many (but not all) coronaviruses [32], [27], inducing the fusion with the virion membrane and forming a pore for the passageway of viral material inside the cytoplasm.
Cell receptor binding through the spike protein
Concerning the interaction with ACE2, the binding surface involves the RBD motif and it appears somewhat altered in SARS-CoV-2S with respect to other coronaviruses, displaying a much higher (about 6–20-fold) affinity than what reported for other viral Spike proteins (K D ~ 15 nM, as from [33], K D ~ 5 nM, as from [8]).
Molecular modelling studies suggested that the higher affinity of SARS-CoV-2 RBD reflects the substitution of Leu472 (present in SARS-CoV) with Phe486 (in SARS-CoV-2), allowing stronger van der Waals interactions with both Leu79 and Met82 of ACE2. Moreover, SARS-CoV-2S displays a Lys417 (which is Val404 in SARS-CoV), favouring a tighter association through a salt bridge formation with Asp30 of ACE2 [8], [34], [35].
Interestingly, low molecular weight heparin, which seems to induce a structural change of the S1 RBD, likely affects the interaction of RBD with ACE2 [36], bringing about an affinity decrease of the S protein for ACE2 and partially impairing the viral invasion in lung and small intestine epithelial cells, where ACE2 is mostly expressed [37].
Host proteases
FURIN: Structure-Function
Furin (EC 3.4.21.75), also named PACE or PCSK3, is the prototype of subtilisin-/kexin-like proprotein convertase (PCSKs), encoded by a transcription unit in the upstream region of c-fes/fps proto-oncogene and it is ubiquitously expressed in eukaryotic cells and tissues [10].
Furin is a type I transmembrane highly specific endoprotease which cleaves the precursor forms of many secreted proteins during their transport along the constitutive secretory pathway. Notably, furin has been identified as an activating protease for fusion surface glycoproteins of a broad range of viruses [10], displaying a broad optimum pH ranging between pH 5 and pH 8 [14] .
Its large luminal/extracellular region has an overall homology with similar regions of other members of proprotein convertase (PC) family. The multi-segmented pro-precursor (794 amino-acid long) is formed by:
- (i) the N-terminal signal peptide,
- (ii) the pro-domain (which is autocatalytically removed at pH 6.5),
- (iii) the subtilisin-like catalytic domain,
- (iv) the P-domain (which modulates pH and calcium requirements),
- (v) the Cys-rich domain,
- (vi) the transmembrane anchor domain, and
- (vi) the C-terminal cytoplasmic domain (which controls the localization and sorting of furin in the trans-Golgi network (TGN)/endosomal system) (Fig. 4 A).

Schematic representation of the furin structure. (A): Schematic representation of the domains of furin. Each domain is represented by different shape and color and is defined by arabic numbers listed above them. Asp153 (D), His194 (H), Ser368 (S), are the amino acid residues that form the catalytic triad of furin; Asn295 contributes to the oxyanion hole. (B): Crystal structure of human furin (PDB ID: 5JXG) [40]. The catalytic domain of furin is shown with its surface in gold and the P-domain in green. The amino acid residues of the catalytic triad and of the binding sites of furin to the viral S protein are displayed. (C): Crystal structure of mouse furin in complex with the inhibitor Dec-Arg-Val-Lys-Arg-CMK (PDB ID: 1P8J) [53]. Furin is shown in light blue. The inhibitor is displayed in black sticks with nitrogen in blue and oxygen in red. The figures of panels B and C were drawn using the UCSF Chimera software [178]. For details, see the text. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Furin shuttles between the TGN and the cell membrane through the endosomal system, it is synthesized as a multi-segmented zymogen (100 kDa) (Fig. 2), and it is autocatalytically cleaved into the mature form (70 kDa) in the TGN. Furin ectodomain can be shed, and the soluble form also retains its catalytic activity in vivo and in vitro [38] . This essential prodomain has a crucial role in the folding, activation and transport, and thus in the regulation, of furin activity through cell specific compartments.
Furin catalytic site
Like other serine proteases, furin is characterized by a catalytic triad, made up of Ser368, His194, and Asp153 whereas Asn295 contributes to the oxyanion hole [39] (Fig. 4B). Interestingly, the substrate-free enzyme is essentially inactive since the catalytic Ser368 residue is flipped over with respect to the active conformation. Substrate binding triggers the conformational change of Ser253, which brings about a flipping of Ser368 from the inactive to active conformation [40] (Fig. 4B).
The high specificity of furin proteolysis occurs at the C-terminal of a multibasic recognition motif, showing Arg-Xxx-Arg↓Xxx and Lys-Arg↓Xxx as preferred consensus sequences, often containing an additional Arg residue in the P4 position [10]. Unlike trypsin and chymotrypsin, the substrate-binding pocket is rigid and rich of negative charges, e.g. Glu 257 and Asp153 at the S1 and S2 substrate recognition sites, respectively.
The rigidity of the substrate-binding pocket envisages a high selectivity for substrates, which indeed must properly fit inside the recognition site interacting with (at least) 6 residues forming the minimum binding surface [41].
The substrate Arg residue is bound to the Glu 257 side chain of the S1 site, while the positively N-terminal charged residue (i.e., Lys) is bound to the Asp153 residue of the S2 site (Fig. 4B). In particular, Asn133 is a key component of the S5 substrate binding pocket together with Glu236 [41], while the S4 site involves two additional negatively charged residues (i.e., Asp236 and Asp 264) also present at the buried interface [42], [41] (Fig. 4B).
The enzymatic processing of S protein by furin
Although the contribution of furin to SARS-CoV-2 S activation is not clear yet, its role has been demonstrated in many coronaviruses [43]. Furin has been recognized important in mediating MERS-CoV S entry, with a prominent role in the two-steps activation of the spike protein [9], [32].
Beside a canonical furin-like cleavage site, which is shared with other CoVs (i.e., HCoV-OC43, MERS-CoV, and MHV-A59) [13], the SARS-CoV-2 S protein displays at the S1/S2 site a 4 amino acids insertion which provides a minimal furin cleavage site (Pro-Arg-Arg-Ala-Arg689↓Ser-Val) and appears to create a larger S protein-furin interface, showing both positively charged residues (such as Arg185, Arg193, Lys261 and Arg298) (Fig. 3C) and negatively charged ones of the catalytic site.
It is supposed that furin attacks this cleavage site during virus egress [32], facilitating the S protein priming and providing a gain-of-function to the SARS-CoV-2 for efficient spreading in the human population, as compared to other b lineage of β-coronaviruses. Therefore, the inhibition of such a process indeed might represent the basis for a successful antiviral strategy.
Furin inhibitors
Because of the key role of furin in cancer and several infectious diseases, including Human Immunodeficiency virus (HIV) and coronaviruses [44], [14], its suitability as a therapeutic target has raised significant interest for several years. Up to now various compound classes have been identified as promising lead compounds for drug development [45] (see Table 1 ).
Table 1
Inhibitors of Furin and their related applications.
| Inhibitor | Ki (pM) | Application | Refs. |
|---|---|---|---|
| α1-PDX | 600 | Prevents the processing of HIV-1 gp160 and measles virus-Fo in vitro. | [38], [46], [47] |
| Limits joint inflammation when delivered with adenovirus into the joint of arthritic mice. | [176] | ||
| Dec-Arg-Val-Lys-Arg-CMK | 1000 | Topical uses in the treatment of HPV skin infections. | [53], [57], [177] |
| Antiviral activity against HIV, HBV, influenza A, EVD, Chikungunya virus and ZIKV, JEV. | [44], [55], [56], [58] | ||
| Inhibits processing of MERS-S protein in infected cells. | [32], [59] | ||
| Block SARS-CoV-2-S processing in rhabdoviral particles. | [19] | ||
| Phac-Arg-Val-Arg-Amba | 810 | Reduces FPV propagation in a long-term infection test. | [61] |
| Suppresses the activation of HA0 in fowl plague virus in cell-based assays. | [61] | ||
| MI-1148 | 5.5 | Protective effect against H5N1 and H7N1 HPAIV, CDV, and RSV in cells. | [42], [63] |
| MI-1554 | 8.5 | Prevents the proteolytic activation and replication of RSV in cells. | [62], [63] |
Non-peptidic inhibitors
A variety of approaches has been proposed to inhibit furin activity and some protein-based inhibitor variants have been engineered to develop powerful inactivators of furin. Among them, the α-1 antitrypsin Portland (α1‐PDX), a variant of the naturally occurring serine protease inhibitor α-1 antitrypsin, is able to efficiently inhibit furin (almost irreversibly due to its strong affinity) and to prevent the processing of HIV‐1 Env and measles virus F in vitro [38], [46], [47] (see Table 1).
Similarly, peptides have been derived from the cleavage site of influenza A virus hemagglutinin, where polyarginines compete with natural furin substrates [48], [49].
Furthermore, the crystal structure of furin allowed the targeted modelling of non-peptidic inhibitors, such as streptamine-based compounds. Upon addition of guanidine residues, streptamine derivatives mimic the cationic furin cleavage site and inhibit its enzymatic activity in vitro in the nanomolar range [50].
Notably, a 2,5-dideoxystreptamine-based small molecule inhibitor was found to interact with furin showing an unusual binding mode, different from a peptide-based substrate, which inhibits furin with K i = 46 nM. In particular, one molecule of inhibitor anchors at the S4 pocket of the enzyme, directly interfering with the conformation and function of the catalytic triad, while a second molecule shows weaker binding and interacts with a distant, less conserved region of furin [51].
Peptidic inhibitors
Several effective synthetic furin inhibitors have been developed, most of which exhibit multi-basic peptidyl moieties mimicking the substrate sequence [52], [49]. To increase their stability, and thus their efficiency, several approaches have been applied.
In this regard, the most powerful peptidomimetic furin inhibitors were based on the prototypical compound dec-Arg-Val-Lys-Arg-CMK (where dec refers to decanoyl group and CMK to chloromethyl ketone) (Fig. 4C and Table 1), by coupling appropriate multi-basic substrate sequences to a P1 arginyl chloromethyl ketone group [53].
The addition of the CMK moiety has proven useful as it irreversibly alkylates the His194 residue in the active site of furin, blocking its activity [53] (see Fig. 4C and Table 1); dec-Arg-Val-Lys-Arg-CMK has also good cell permeability properties, which in turn enhance its efficacy [54]. Dec-Arg-Val-Lys-Arg-CMK is effective in the reduction of hepatitis B virus (HBV) replication by inhibiting furin-mediated processing of the hepatitis B e antigen (HBeAg) precursor into mature HBeAg [55] .
It was also reported that it is able to inhibit furin-mediated cleavage and fusion activity of viral glycoproteins, and acts as an antiviral agent against different viruses, including HIV [44], Chikungunya virus [56], chronic HBV, influenza A, Ebola virus infection [55] and papilloma virus [57].
Recently, the efficacy of dec-Arg-Val-Lys-Arg-CMK as an anti-flavivirus agent (against Zika virus and Japanese encephalitis virus) has been demonstrated in both mammalian cells and mosquito cells with significant antiviral activities in terms of the reduction in virus progeny titre, in viral RNA and protein production [58].
In the context of the coronavirus infection, dec-Arg-Val-Lys-Arg-CMK inhibited the processing of MERS-S protein, in a concentration-dependent manner, and had no effect on SARS-CoV-S expression, as expected from substrate specificity [32], [59]. Noteworthy, this furin inhibitor has been shown to block SARS-CoV-2 S processing at the S1/S2 site [60], strengthening the idea that indeed furin is involved in the S1/S2 priming.
Nevertheless, as a result of the cytotoxicity of CMK-based inhibitors and of the instability of the CMK moiety, the potential therapeutic applications of these molecules remain limited to topical applications, such as the treatment of papillomavirus (HPV) skin infections [53], [45].
However, the incorporation of amino acid analogues, such as D‐amino acids, decarboxylated P1 arginine mimetics, and 4-amidinobenzylamide (Amba), increased the stability of peptide-derived furin inhibitors. The most powerful compound, Phac-Arg-Val-Arg-Amba (Phac being a phenylacetyl group), inhibits recombinant furin with K i = 0.81 nM [61].
Despite its excellent activity in vitro, Phac-Arg-Val-Arg-Amba showed reduced potency (IC50 ~ 10 μM), in a cellular assay, as an inhibitor of the cleavage of the fowl plague hemagglutinin of the H7 subtypes of the avian influenza viruses, possessing a multi-basic furin cleavage site; this limited efficacy might be related to a reduced ability of this inhibitor to target intracellular furin.
The same authors also showed that this inhibitor was able to reduce feline panleukopenia virus propagation in a long-term infection test (25 μM of inhibitor over a period of 72 h, see Table 1).
Another powerful inhibitor of furin is a peptido-mimetic (i.e., 4-(guanidine methyl)-phenyl acetyl-Arg-Tle-Arg-4-Amba (MI-1148), where Tle refers to tert-leucine, also named 3-methylvaline or tert-butylglycine, see [42]), which displays a 300-fold increase of the affinity (=5.5 pM, see [42]) for the substrate pocket, essentially blocking the furin activity (see Table 1).
Inhibitor MI-1148 was shown to have a significant protective effect against anthrax and diphtheria toxin and to be active against H5N1 and H7N1 avian influenza viruses (HPAIV) and canine distemper viruses (CDV) propagation in cell culture. Interestingly, MI-1148, its P2 Lys analogue MI-1554 (4-guanidinomethyl-Phac-Arg-Tle-Lys-4-Amba, K i = 8.5 pM, see [62]) and several cyclic hexapeptide derivatives were recently tested as inhibitors of the proteolytic activation and replication of respiratory syncytial virus in cells [63] (see Table 1). Significant antiviral activity was found for both MI-1148 and MI-1554 linear inhibitors, whereas a negligible efficacy was determined for the cyclic derivatives.
The authors have speculated that the specific chemical structure of MI-1148 and other close linear analogues might enable enhanced cellular uptake, providing improved intracellular inhibitory potency.
TMPRSS2: Structure-function
A key discovery in understanding the mechanism of SARS-CoV-2 infection concerns the role of the androgen-responsive transmembrane serine protease 2 (TMPRSS2), a cell-surface protein that has been shown to facilitate the SARS-CoV-2 entry in the human airways by cleaving the viral Spike (S) protein [22], [23], [19].
TMPRSS2 (EC 3.4.21.109) is preferentially expressed in several epithelial tissues, such as prostate, kidney, colon, small intestine, pancreas, and lungs [19], [22], [23], [64].
The physiological role of TMPRSS2 is unknown yet, even though it has been demonstrated that the expression of TMPRSS2 in lung cancer cell line A549 and prostate cancer cell is androgen-dependent [22], [65], [66]. TMPRSS2 is also expressed in the cardiac endothelium, kidney, and digestive tissues, which indeed are target tissues for SARS-CoV-2 infection.
As a matter of fact, among the clinical complications of COVID‐19 myocardial and acute kidney injuries are reported together with gastrointestinal symptoms [67], [68], [69]. Furthermore, since TMPRSS2 is also expressed in microvascular endothelial cells of the blood vessels, the SARS‐CoV‐2 virus may bring about endothelial dysfunction with associated thrombosis [70], [68].
Notably, epidemiological studies, carried out in several countries (i.e., China, Italy and the United States), suggest that the incidence and severity of the diagnosis of COVID-19 and other TMPRSS2-dependent viral infections are higher in men than in women. In this regard, the role of TMPRSS2 in prostate cancer and the androgen-dependent TMPRSS2 expression has led to speculate that the prevalence of COVID-19 cases in men may possibly be related to TMPRSS2 [71].
TMPRSS2 gene is located on human chromosome 21q22.3 and includes 14 exons [72], [23]. A feature of the TMPRSS2 gene is that several androgen receptor elements (AREs) are located upstream of the transcription start site and the first intron of the TMPRSS2 gene [22], [65], [73], [66]. TMPRSS2 gene encodes for a predicted protein of 492 amino acids that belongs to the type II transmembrane serine proteases (TTSPs) family [74], [75].
The TTSP family is characterized by: (i) the N-terminal transmembrane domain, (ii) the C-terminal extracellular serine protease domain of the chymotrypsin (S1) fold that contains the catalytic His, Asp, and Ser residues, and (iii) the “stem region” that contains a mixture of one to eleven protein domains of six different types (Fig. 5 A).
TTSPs are synthesized as single-chain precursor and their activation produces a two-chains form which is stabilized by a disulfide bridge, anchoring the active form to the cell membrane. Nineteen human TTSPs have been identified and may be divided into four subfamilies, namely Hepsin/TMPRSS, Matriptase, HAT/DESC, and Corin [74], [75].

Schematic representation of the TMPRSS2 structure. (A): Schematic representation of the domains of TMPRSS2. Each domain is represented by different shape and color and is defined by arabic numbers listed above them. His296 (H), Asp345 (D) and Ser441 (S) are the amino acid residues that form the catalytic triad of TMPRSS2. (B): The three-dimensional model of TMPRSS2 was built according to [90]. The catalytic domain of TMPRSS2 is shown with its surface in gold, the SRCR domain in cyan and the LDL domain in pink. The amino acid residues of the catalytic triad and of the predicted active site are displayed. (C): Three-dimensional model of TMPRSS2 in complex with the standard inhibitor camostat mesylate [90]. The catalytic triad (i.e., His296, Asp345, Ser441) and the predicted interactions of camostat with the active site residues (i.e., Asp187, Asn346, Cys348, and Asn450) of the human serine protease are shown. TMPRSS2 is shown in tan. The inhibitor is displayed in black sticks with nitrogen in blue, and oxygen in red. The figures of panels B and C were drawn using the UCSF Chimera software [178]. For details, see the text. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
TMPRSS2 catalytic site
To date, the three-dimensional structure of human TMPRSS2 protein has not been solved, since it displays a high percentage of coiled structure, a theoretical isoelectric point of 7.42, and a high surface hydrophobicity [72], [76]. Full length structural models show that TMRPSS2 has a considerable structural homology with the serine protease hepsin (PDB ID: 1Z8G) [77].
In detail, the TMPRSS2 structural model (Fig. 5A) shows:
- (i) the N-terminal region, weakly structured as a low density lipoprotein (LDL)-receptor class A,
- (ii) putative Ca2+ binding residues (i.e., Asp134, His138, Asp144, Glu145, and Ile256) on a loop linking the N-terminus of the protein with the Scavenger Receptor Cysteine-Rich (SRCR)-domain,
- (iii) the SRCR domain, composed of an α helix and multiple anti parallel β sheets stabilized by two disulfide bonds between Cys172-Cys231 and Cys185-Cys241,
- (iv) the C-terminus, with typical structural features of chymotrypsin family serine proteases, characterized by the His296, Asp345, and Ser441 catalytic triad and the substrate binding sites (i.e., Asp435, Ser460 and Gly462) sandwiched between two β barrels, each being composed of six strands of nearly equal size [78], [76].
The globular conformation of the domains is likely stabilized by four disulfide bonds between Cys244-Cys365, Cys281-Cys297, Cys410-Cys426, and Cys437-Cys465 [78], [76] (Fig. 5).
The catalytic mechanism of the TMRPSS2 involves a catalytic triad of three amino acids, namely Ser441 (nucleophile), Asp345 (electrophile), and His296 (base). The catalytic reaction hydrolyzes the substrate by a two-steps mechanism, that is (a) the acylation step, which involves the formation of a covalently linked enzyme-peptide intermediate and the loss of a peptide fragment, followed by (b) the deacylation step, characterized by a nucleophilic attack on the intermediate by water, leading to the hydrolysis of the peptide [79], [75]. To date, TMPRSS2 substrate specificity and catalytic properties have not been well characterized [75].
The TMPRSS2 cleavage of S protein
TMPRSS2 is involved in the proteolytic activation of influenza virus (cleaving the viral hemoagglutinin A) and coronavirus (cleaving the viral Spike (S) protein), thus contributing to the virus invasion of human airways [80], [81], [66].
Not only SARS-CoV-2 but also other types of coronaviruses and influenza viruses (such as the SARS-CoV responsible for the 2003 SARS outbreak and the influenza H1N1 responsible for the 1918 and 2009 influenza pandemics) depend on TMPRSS2 for activation of their spike S protein and the consequent cellular invasion [82], [83], [19], [71].
In vitro and in Vero cells studies demonstrated that the inhibition of TMPRSS2 protease activity by molecules, such as Camostat mesylate, partially inhibits the entry of SARS-CoV-2 into the lung epithelial cells [84], [19], [15]. Furthermore, TMPRSS2-deficient mice had minimal initial infection when infected with specific influenza A virus strains, SARS-CoV and MERS-CoV, respectively, showing an attenuated disease progression, as compared to wild type control mice. This protection indeed is likely due to the inhibition of the proteolytic activation of progeny virus and consequently inhibition of virus spread along the respiratory tract [85], [86], [87], [88].
Of note, although the SARS-CoV-2 may use cathepsin B/L or TMPRSS2 for proteolytic priming (see Fig. 2), only TMPRSS2 is essential for viral spread and pathogenicity (cathepsin B/L activity being dispensable) [89], [88], [90]. In this regard, it has been demonstrated that the transient expression of TMPRSS2 in Vero cells favors the cathepsin-independent entry of SARS-CoV-2. Furthermore, the pre-treatment of human Caco-2 colon and human airway cells with TMPRSS2 inhibitors reduces the entry of SARS-CoV-2 [84], [15], [19].
TMPRSS2 cleaves the S protein of coronavirus at two potential sites (Arg689/Ser690 and Arg815/Ser816 at the S1/S2 and the S2′sites, respectively) (Fig. 3C and 3D) generating two distinct fragments of the S protein [19], [76]. Selected docking poses for the complex between TMRPSS2 and SARS-CoV-2 S protein show that both cleavage sites of the viral S protein (i.e., S1/S2 site and S2′, see Fig. 3B) interact with one of the β barrel of the catalytic domain of TMPRSS2. In detail: (i) at the first cleavage site (Arg685/Ser686), the TMPRSS2 His296 establishes a hydrogen bond with the viral S protein residue Arg682; (ii) at the second cleavage site (Arg815/Ser816), His296 and Ser441 of TMPRSS2 form hydrogen bonds with Pro809, Lys814 and Ser810 of the S protein. A hydrogen bond also occurs between Ser810 of the S protein and the Ser460 of TMPRSS2 at the substrate binding site (Fig. 5B). Furthermore, the S protein Ser810 forms a hydrophobic interaction with the His296 of TMPRSS2 at the catalytic site [76] (Fig. 5B).
Since the key residues of TMPRSS2 interact with amino acids flanking the cleavage site of S protein, it has been suggested that, upon interaction with TMPRSS2, the protein S may undergo a conformational change needed for the fine positioning of the cleavage site (i.e., residues Arg685/Ser686 and/or Arg815/Ser816) into the active site cleft [76].
TMPRSS2 inhibitors
Alpha-1-antitrypsin (A1AT), 4-(2-aminomethyl)-benzene-sulfonyl fluoride (AEBSF), Camostat mesylate, Nafamostat and Bromhexine hydrochloride are the best experimentally validated inhibitors of TMPRSS2, as it results from in vitro cell experiments and computational approaches [64], [66], [91], [19], [20], [92].
A1AT is a small protein, synthesized in the liver and present in plasma at high levels (0.9 g/L), its concentration displaying a six-fold increase under acute inflammation [93]. In the lungs, A1AT acts as a protective protein of the tissue damage and inflammatory response blocking the action of proteases involved in the cleavage of several structural proteins and processing several mediators of the innate immune response. It has been speculated that A1AT may reduce the pathogenicity of COVID-19 both by inhibiting the alveolar inflammatory response and by acting as a TMPRSS2 inhibitor, blocking the entry of the virus into the host cell [91].
AEBSF is a small molecule that blocks nonspecifically the protease activity (including TMPRSS2), occupying the S1 pocket of trypsin-like serine proteases and leading to a covalent sulfonylation of the active site [94], [91]. It brings about a decrease in the levels of both H1N1 and H7N7 nuclear proteins within the lung tissue of mice infected with influenza; further, AEBSF partially inhibits also the fusion of the mouse hepatitis coronavirus [95].
Bromhexine hydrochloride (BHH) is a drug used as a mucolytic and cough suppressant that shows specific inhibition of TMPRSS2 (IC50 = 0.75 μM). Given that BHH is an FDA-approved drug with no significant adverse effects, it could be used for treatment of coronavirus infections as an inhibitor of TMPRSS2 [64], [66], [92].
Camostat mesylate is considered the standard inhibitor of TMPRSS2 and it has been demonstrated that it is able to inhibit the proteolytic activity of TMPRSS2 even at the lowest concentration of 100 nM, thus reducing the probability of SARS-CoV-2 penetration in cell experiments in vitro [84], [19], [91] (see Fig. 5C). Similarly, the Nafamostat, a structural analog of Camostat, blocks SARS-CoV-2 infection of human lung cells with 15-fold higher efficiency than Camostat mesylate; this makes it a good compound to enter clinical trials for COVID-19 treatment [20].
A structural model of TMPRSS2-Camostat complex suggests that the predicted active site of the TMPRSS2 consists of the amino acid residues Asn146, Arg147, Cys148, Val149, Arg150, Leu151, Asp187, Met188, Tyr190, Ile221, Tyr222, Lys223, Asn368, Pro369, Gly370, Met371, Lys449, Asn450, Ile452, and Trp454 [76], [92].
Camostat interacts with the TMPRSS2 Val28, Asp440, Thr459, Ser460, Trp461, Tyr474 by van der Waals interactions and establishes seven hydrogen bonds with four key residues of the protease active site (i.e., Asn146, Cys148, Asp187, and Asn450) [76].
In detail, Asn146 forms an arene cation and backbone acceptor H-bonds, while Cys148 forms an arene-H bond with the benzene ring of the ligand. Asn450 forms two hydrogen bonds with the anhydrous carbonyl oxygen, with side chain acceptor and backbone acceptor.
Asp187 forms an acidic hydrogen bond with the primary amine and a sidechain donor hydrogen bond with the secondary amine of the ligand [76]. In addition, different groups of Camostat mesylate form strong hydrogen bonds with two amino acid residues of the TMPRSS2 catalytic triad (i.e., His296 and Ser441) (Fig. 5C).
Nafamostat and Camostat mesylate bind in the same pocket of TMPRSS2 and in the same way. On the other hand, due to its small structure, BHH binds the active site of TMPRSS2 with fewer hydrogen bonds and more hydrophobic interactions, as compared to Camostat mesylate and Nafamostat, establishing hydrophobic interactions with the TMPRSS2 His279, Val280, Cys281, and His296 [92] (Fig. 5C).
The binding energy of TMPRSS2 with Camostat mesylate, Nafamostat and Bromhexine hydrochloride is of −7.94 kcal/mol, −7.21 kcal/mol, and −5.96 kcal/mol respectively. Similarly, the inhibition constant (K i) of Camostat mesylate, Nafamostat and Bromhexine hydrochloride are 1.51 µM, 5.17 µM and 43 µM, respectively [92].
A recent analysis, based on the combination of a ligand-based pharmacophore approach and a molecular docking-based screening, allowed to identify 12 potential natural inhibitors of TMPRSS2. Among these drug-like compounds, the geniposide, that is the major iridoid glycoside of gardenia fruit (IUPAC name: methyl (1S,4aS,7aS)-7-(hydroxymethyl)-1-[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1,4a,5,7a-tetrahydrocyclopenta[c]pyran-4-carboxylate), showed the highest docking score of −14.69 kcal/mol [76]. Geniposide forms 10 hydrogen bonds with the active site residues of the receptor protein. Among these H-bonds, five TMPRSS2 amino acid residues (i.e., Asn146, Arg147, Arg150, Lys449, and Asn450) are side chain acceptor, and two residues (i.e., Asn146 and Arg147) are concurrently backbone acceptor and donor. It is known that the geniposide is an inhibitor of 5-lipoxygenase [96] and it has anti-angiogenic activity [97] as well as anti-asthmatic properties [98].
Inhibitors for host proteases as therapeutic platform
As it happened in the past for the HIV, HCV, influenza viruses, SARS-CoV and other etiological agents, today a great effort is being devoted to the SARS-CoV-2 pandemia, showing its own peculiar protein targets. In the absence of an effective vaccine, inhibitor repurposing or de novo drug design may offer an effective strategy to combat the alarming SARS-CoV-2 pandemia.
As reported above, an interesting potential antiviral strategy concerns the inhibition of ACE2-dependent viral entry, interfering with the cleavage of SARS-CoV-2 spike protein to arrest the viral propagation.
Therapeutic platform for furin
Within the lineage b of β-coronaviruses, SARS-CoV-2 has revealed a dominant importance of furin, which was not observed before, suggesting that furin inhibitors may participate to halt virus propagation. However, despite considerable evidence of in vitro and in vivo efficacy of furin inhibitors, there is very limited clinical trial evidence to support or reject the use of these compounds in a therapeutic context.
Moreover, one of the greatest limitations for the development of furin inhibitors as therapeutic agents has been related to potential health consequences. In this regard, since furin-like enzymes are involved in a multitude of cellular processes [99], an important issue would be to avoid systemic inhibition that may lead to relevant side effects.
While many of the inhibitors, described above, strongly reduce furin activity, most of them also inhibit other PCSKs, recognising the same or similar polybasic cleavage sites [45]. Although a selective inhibition of individual PCSKs can be achieved, systemic long‐term inhibition will most likely have detrimental effects. Therefore, one crucial point in the therapeutic application of furin inhibitors is to limit the systemic inhibition that may lead to some adverse effects.
It must be stressed that, differently from viral targets, furin (like any protein hijacked by the virus system) is an attractive target as it is not expected to develop drug resistance. On the other hand, as for Ebola virus, although a furin cleavage site has been clearly demonstrated, blocking the furin-mediated cleavage of glycoprotein does not result in a reduction of viral replication under cell culture conditions, suggesting that the inhibition of furin activity may not always produce beneficial effects [100]. Therefore, to encourage further developments of furin inhibitors additional proofs-of-concept are required at each step of the different infectious processes.
An additional important aspect concerns the assessment of cell penetration properties for the developing inhibitors. Since furin localizes both in intracellular compartments (i.e., secretory pathway, endosomal pathway) and at the cell surface, inhibitors will have to reach both destinations to ensure full cleavage inhibition. Of note, several furin inhibitors (e.g., polyarginine and dec-Arg-Val-Lys-Arg-CMK) exhibit good cell permeability properties, which in turn enhance their efficacy [101], [54] .
A possible strategy to limit viral infections is the restriction of the trafficking of furin to trans-Golgi network or to early Golgi compartments, where the pro-protein convertase remains inactive [13]. In this regard, streptamine derivatives may be particularly promising for targeted therapy, since the positioning of the guanidyl substituents leads to a localisation of the inhibitor into distinct subcellular compartments, such as endosomes or the Golgi complex [102].
Overall, despite the crucial role of furin, it is possible that acute inhibition over a limited time interval may lead to vastly beneficial effects in tackling viral infections, especially in the case of a substantial ineffectiveness of vaccines or other drugs. Future studies will have to better define short- and mid-term toxicity profiles and thus to establish the pharmacological safety of this type of intervention. Although the therapeutic application of furin inhibitors may show several pitfalls, it is certainly a potential treatment option against SARS-CoV-2 that should be further pursued [103].
Therapeutic platform for TMPRSS2
Although Camostat mesylate blocks COVID-19 entry into the cell by inhibiting the cellular host TMPRSS2 (see above sections), more testing is required before it can possibly be labelled as an effective therapy for the treatment of COVID-19.
Considering the central role of TMPRSS2 in activating SARS-CoV-2 and other respiratory viral infections, to interfere on its expression or on its activity indeed could represent a promising approach to treat respiratory COVID-19. Therefore, the knowledge of the amino acids that make up the active site of TMPRSS2 and the cleavage site of SARS-CoV-2 favors the targeted design of efficient drugs against COVID-19.
The structural information, obtained by computational analysis, combined with experimental in vitro and in vivo validation, represents the basis for designing and discovering new protease inhibitors to be used for preventing the entry of SARS-CoV-2 into human host cells.
Among potential anti-COVID-19 drugs we report the phytochemical geniposide; this TMPRSS2 inhibitor should be validated for treatment of coronavirus infection as it does not show toxicity in humans, since it has been reported that geniposide cannot cross the blood–brain barrier and it is not absorbed by the gastrointestinal tract [90]. In addition, geniposide protects against sepsis-induced myocardial dysfunction by activating AMPK which suppresses myocardial reactive oxygen species (ROS) accumulation [104].
Two protease inhibitors (i.e., camostat, at a concentration of 0.1–10 µg/ml, and nafamostat, at a concentration of 0.01–1 µg/ml) inhibit the coronavirus replication in human airway epithelial cells, displaying an additive effect in combination with interferon [105].
The antiviral action of nitric oxide (NO) has been reported for the treatment of several DNA and RNA virus families [106]. Therefore, a novel approach to arrest SARS-CoV-2 life cycle could be targeting cysteines of TMPRSS2 through a covalent attachment of a nitrogenous group (–NO). As NO and NO donors are able to nitrosylate thiol group, inhaled NO in COVID-19 patients could potentially be an effective therapy [107].
Virus proteases
The polyproteins encoded by ORF1a and ORF1b are auto-catalytically cleaved into 16 different non-structural proteins (nsp) by the two viral proteases (i.e., Mpro and PLpro). The N-terminal part, which is cleaved by PLpro, contains three proteins, namely nsp1 and nsp2, which help modulating the host response, and nsp3.
PLpro activity also plays a key role in other functions, such as the deubiquitination of host polyubiquitin chains and the formation of the viral double-vesicles membranes (see Fig. 1).
The rest of pp1a and pp1ab are self-cleaved by Mpro, giving rise to 13 proteins which, except for Mpro itself, play different roles in RNA replication (i.e., helicase, proofreading exoribonuclease, endoribonuclease and methyltransferase activities).
It is worth citing here that the polyprotein pp1ab, which is encoded when ribosomal frameshift occurs, also contains the RNA-dependent RNA replicase, a common target for anti-viral inhibitors.
Main protease (3CL)
Sequence similarities in coronaviruses Mpro
Besides SARS-CoV-2, only six other human coronaviruses have been identified so far and belong to two distinct genera, namely (a) Alphacoronavirus (i.e., (i) HCoV-NL63 and (ii) HCoV-229E) and (b) Betacoronavirus (i.e., (i) HCoV-OC43, (ii) HCoV-HKU1, (iii) the severe acute respiratory syndrome SARS-CoV, and (iv) the Middle East respiratory syndrome MERS-CoV) [108].
The multiple-sequence alignment of the main proteases from the seven human coronaviruses (Fig. 6 A) shows that the active site (i.e., residues Thr24-Leu27, His41-Tyr54, Phe140-Cys145, His163-Pro168) is highly conserved in all the coronavirus Mpros, clearly indicating its specificity.
Interestingly, from a pairwise alignment, SARS–CoV-2 Mpro shows more similarities to its SARS-CoV homologue (96% identity and 99% similarity) than with any of the other six Mpros from human coronaviruses (average identity 50%, average similarity 64%). Indeed, only very few residues in SARS-CoV-2 are substituted with respect to the SARS-CoV counterpart, that is Thr35Val, Ala46Ser, Ser94Ala, Lys180Asn, Ala267Ser, and Thr285Ala.
All these mutations are located in poorly conserved regions on the surface of Mpro with the exception of Ser46, which is located in the proximity of the active site entrance; the Ala267Ser mutation is also observed in the HCoV-NL63 strain. Although such a small structural change would be not expected to substantially affect the binding of small molecules to the binding sites, structural and molecular modelling studies show that Ala46Ser may have a relevant effect on the shape and flexibility of the Cys44-Pro52 loop at the active site entrance (see below), where substitutions occur quite often during the evolution of viral Mpro proteins [109].

Analysis of sequence and structure of Mpro (3CL) protease. (A): Multi-alignment of Mpro homologues from the seven known human coronaviruses: SARS-CoV-2 (Uniprot ID: P0DTD1), SARS-CoV (P0C6X7), HCoV-OC43 (P0C6U7), HCoV-KU1 (P0C6U3), MERS-CoV (V9TU05), HCoV-NL63 (P0C6U6), HCoV-229E (P0C6U2). Secondary structure references were taken from SARS-CoV-2 Mpro (PDB: 6M2Q). (B): Three-dimensional structure of apo SARS-CoV-2 Mpro (PDB: 6M2Q). The structural domains I, II and III of Mpro are colored in light blue, orange and green, respectively. The localization of the protease active site (i.e., residues Cys41 and His145) and the “N-finger” are indicated by arrows. (C): SARS-CoV-2 Mpro complexed with N3 peptide (PDB: 6LU7). Mpro structure is represented by molecular surface colored with the same scheme used in panel B. Subsite pockets S1 and S2 are explicitly indicated. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Structural features of Mpro proteases in SARS-CoV-2
In the mature active form, Mpro (EC 3.4.22.69) is found as a homodimer. Each protomer is formed by three structural pseudo-domains, namely two antiparallel-β-barrel domains (domains I and II, residues Phe8-Tyr101 and Lys102-Pro184, respectively), and a five-fold antiparallel-α-helix domain (domain III, residues Thr201-Thr303) [110] (Fig. 6B).
The Mpro enzyme resembles the structure of cysteine proteases, although in the active site the third catalytic residue is missing; thus, the active site is formed by the catalytic dyad His41-Cys145, which forms the “oxyanion hole” together with the main-chain amides of Gly143 and Ser144 [111] (see Fig. 6B).
The His-Cys dyad is highly conserved among the coronavirus proteases and shows significant structural homologies with the 3chymotrypsin protease (3Cpro) of rhinoviruses, which, however, contain a catalytic triad composed of His, Cys, and Glu or Asp [110], [112].
In this context, it is also worth noting that, while the autocatalytic process of viral polyproteins is performed by both Mpro and PLpro, the two proteinases have instead significantly different features. As a matter of fact, mature PLpro is a multifunctional protein composed by an N-terminal ubiquitin-like domain (not involved in the catalytic function) and a cysteine protease core domain, not sharing any structural feature with Mpro [113].
The Mpro active site has different subsite pockets which confer a high substrate specificity for each residue in the recognized cleavage sequence [114], [115]. These specificity pockets are named as S1, S2, S4, S1′ after the residues in position P1, P2, P4, P1′ of the substrate sequence. When bound to coronavirus Mpro, the P4 and P3 residues adopt a β conformation, inducing structural adaptation of the corresponding subsites, while both P1 and P2 side-chains are allocated in the pre‐formed S1 and S2 specificity pockets [115] (Fig. 6C).
For SARS-CoV and MERS-CoV Mpro a direct role in the S1 shaping was found to be played by the protonation of two histidines (i.e., His163 and His172 in SARS-CoV sequence) flanking the S1 pocket [116]. Interestingly, a particularly stable water molecule seems also to play an important role by forming a water-bridge that stabilizes His163 and His172 conformations [117].
Additionally, available crystallographic structures of Mpro from SARS-CoV, SARS-CoV-2 and MERS-CoV reveal that the P1 glutamine side chain may interact with the imidazole side chain of His163 which is located at the very bottom of a hydrophobic pocket (i.e., residues Phe140, Ile141, Leu165, Glu166 and His172) which flanks the S1 subsite (Fig. 6C) [118], [111].
Among the other subsites of coronavirus Mpro, the S2 specificity pocket, formed by residues Leu164, Pro188, Ile51, His41 and Thr47, is large enough to display a high specificity for Leu side chain (Fig. 6C). Molecular modelling also suggested that the flexibility of regions 140–146 and 184–197, which encompass S1 and S2 binding pockets, is crucial to accommodate substrate and analogs [117], [119].
The quaternary structure of Mpro may also contribute to the modulation of enzyme activity; thus, the dimer interface area, which is constituted by the two domains III, seems to play a pivotal role by allowing the ion-pair interaction between residue Glu290 of one protomer and Arg4 of the other one. Moreover, in the mature protein, the “N-finger” (the N-terminal residues) of each monomer forms stable hydrogen bonds with Phe140 and Glu166 of the other protomer, squeezing the outer-wall of S1 and shaping the S1 pocket to become catalytically competent [115], [120], [111], [117].
An additional contribution to the dimer stabilization may originate from the direct interaction between the residues 285 of each protomer (Thr and Ala in SARS-CoV and SARS-CoV-2 MPro, respectively); in SARS-CoV MPro this interaction occurs through a H-bond between the two Thr285 hydroxyl groups. However, this interaction, though tightening the dimer assembly in SARS-CoV Mpro, does not seem to affect the catalytic activity, since it is closely similar for the two Mpros [115], [120].
A structural comparison of SARS-CoV-2 Mpro with other coronaviruses Mpros reveals additional amino acid substitutions (usually located far from the catalytic site, see Fig. 6A), but two of them (namely Lys180Asn and Ala46Ser) are potentially relevant, since they are found in the deep hydrophobic pocket below the active site and in the loop region flanking its entrance (i.e., Cys44-Pro52 loop).
Although Lys180Asn is located too far to directly contribute to substrate binding, it clearly extends the hydrophobic inner region of the site. On the other hand, even though the Ala46Ser substitution is not expected to have significant effect on the binding of small ligand compounds[121], molecular dynamics simulations showed that it increases the flexibility of the Cys44-Pro52 loop modifying the active site entrance and likely playing a role in substrate recruitment [109].
Further, a comparison of differing residues between SARS-CoV and SARS-CoV-2 Mpros, has shown that the Cys44-Pro52 loop and the Phe185-Thr201 linker loop are evolutionarily correlated, so that mutations, occurring in these flexible loops, can render them more stable. Interestingly, molecular dynamics simulations of SARS-CoV and SARS-CoV-2 Mpros also showed that, despite the high sequence/structure similarity, their active site binding cavities have significantly different shape with an overall accessible volume 50% larger in SARS-CoV Mpro than in its SARS-CoV-2 counterpart [109]. Thus, these findings suggest that repurposing SARS-CoV drugs for SARS-CoV-2 may not work well [109].
Overall, several studies proved that the few differences between SARS-CoV-2 Mpro and homologue proteases from other human coronaviruses may have significant effects on both dimer stabilization (residue 285) and on active site plasticity (residues 46 and 180).
PLpro protease
As already reported above, the Mpro is not the only protease that processes the post translational maturation of the viral polypeptide product but there is also a papain-like protease (PLpro) (Fig. 2). Unlike Mpro, SARS-CoV and SARS-CoV-2 PLpro share only 83% sequence identity with substantial variations occurring on the protein surface.
These differences are expected to influence the binding of ligands to the PLpro but not the overall secondary and tertiary structures, suggesting that inhibitors developed for SARS-CoV would possibly work as lead compounds also for the development of SARS-Cov-2 PLpro drugs [1].
The papain-like protease (PLpro) is part of the multi-domain/multi-functional non-structural protein 3 (nsp3) which is highly conserved among coronaviruses. Besides the proper proteinase domain, nsp3 also contains two ubiquitin-like (Ubl) domains, an ADP-ribose-1′-phosphatase domain, a nucleic acid-binding domain and trans-membrane domains [122].
The catalytic domain of PLpro recognizes the tetrapeptide Lys-Xxx-Gly-Gly motif at P4-P1 positions found in-between viral non-structural proteins (i.e., ns1 and 2, ns2 and 3, ns3 and ns4), but it shows a broad substrate specificity at P3 position. SARS-CoV-2 PLpro and SARS-CoV PLpro differ by 54 residues, but the P3 and P4 sites are almost identical.
Viral protease inhibition
An efficient inhibition of a multimeric protease can be gained not only by targeting the active site but also by interfering with the stability of its structure and with the modulation of its activity. As an example of this, we can mention darunavir which blocks dimerization of the HIV-1 protease [123], thus impairing its activity and HIV replication. As previously mentioned, proteases may appear evolutionary distant as differing for their active-site, but still share some important functional properties.
As a matter of fact, in the following sections we will report not only on molecules specifically tailored for the active site of Mpro and/or PLpro, but also on molecules, which, though designed for other types of proteases (such as HIV-1), may nonetheless interfere with non-specific processes required for the activity of SARS-CoV-2 proteases.
Mpro inhibitors
In the following, we will present a discussion on inhibitors for human coronavirus Mpro. First, results for known Mpro inhibitors will be presented and recent findings on natural compounds or repurposed drugs as SARS-CoV-2 Mpro inhibitors will be also thoroughly discussed. It is worth noting that the urgency for an effective treatment against COVID-19 has deeply stimulated the scientific community to focus on the development of SARS-CoV-2 Mpro inhibitors. For this reason, some of the results reported here from literature are based on computational assessments and still lack experimental validation.
Available crystallographic structures and molecular modelling studies put in evidence the general structural and dynamical properties of SARS-CoV-2 main protease in relation to its catalytic function. In this context, co-crystallized structures of Mpro in complex with ligands clearly offer new insights which can be exploited for the development of inhibitors.
The first crystallographic structure of SARS-CoV-2 Mpro (a complex with an N3 inhibitor, PDB ID: 6lu7) was released to public on February 2020 through the Protein Data Bank [124]. N3 inhibitor (N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-L-valyl-N ~ 1~-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-L-leucinamide) was identified, through virtual drug- and high-throughput screenings, as a Michael acceptor inhibitor against SARS-CoV and MERS-CoV Mpros; in particular, N3 forms a covalent bond becoming an irreversible inhibitor of SARS-CoV-2 Mpro (Fig. 6C).
Since then, more than 150 structures of SARS-Cov-2 Mpro complexes with putative inhibitors have been made available, although many of them are not presented and discussed yet in a published research work. Interestingly, the co-crystallized structures showed how the absolute requirements of Gln in P1 can be easily overcome by replacing the amino acid with a lactam group which can form a hydrogen bond with His163 [124], [125], [111].
Moreover, crystallographic structures of Mpro in complex with inhibitors showed how the common β-conformation of residues in position P1-P4 can be easily interspersed by ad-hoc spacers to enhance the half-life of the compound, provided that the overall number of hydrogen bonds with the main-chain of residues forming the enzyme specificity subsites remain constant [125], [97].
While in the last decades a relatively small number of docking and inhibition studies have been performed on the Mpro of the closest taxonomic SARS-CoV (the etiological agent of epidemic in 2002), the extensive production of computational research works on SARS-CoV-2 Mpro is absolutely contingent to the actual historical context that combines a common feeling in fighting the pandemic with a planetary spread of software and hardware technologies suitable for the purpose of molecular docking.
In the following, for each Mpro inhibitor all the best poses of potential inhibitors often showed hydrophobic interactions and formation of hydrogen bonds network will be addressed, this finding being in line with the features of the Mpro active site as described in the first part of the section.
It is important to underly that unfortunately a straightforward comparison among inhibitors cannot be accurate, since the affinity scoring functions (expressed in kcal/mol), obtained by different molecular docking processes, cannot be directly converted in terms of binding free-energy values, as they are significantly dependent on the force-fields and protocols employed.
Over one decade ago, in an interesting anti-SARS drug screening research, 59,363 compounds were docked, 93 were selected for inhibition assays, and finally 21 showed inhibition against SARS-CoV Mpro with IC50 ≤ 30 µM. Similar substructures were found in three databases identifying another 25 compounds that exhibited inhibition with IC50 from 3 µM to 1 mM against SARS-CoV Mpro. The promising compounds were also thouroughly investigated by 3D-QSAR and pharmacophore approaches [126].
In the framework of the actual coronavirus pandemia, selected drugs, like favipiravir, amodiaquine, 2′-fluoro-2′-deoxycytidine, and ribavirin are not classified as protease inhibitors, but they were docked and evaluated as possible inhibitors of SARS-CoV-2 MPro. The amodiaquine showed the best binding energy (Fig. 7 A) (−7.77 kcal/mol), envisaging a high binding affinity, which was attributed to the presence of three hydrogen bonds, hydrophobic interactions between the drug and critical residues of the Mpro as well as to its electrophilicity index, basicity, and dipole moment [127].

Ligands docked in Mpro active site. Examples of ligands docked in the active site: (A) Amodiaquine; (B) Bonducellpin D; (C) Heptafuhalol; (D) Simeprevir; (E) Pitavastatin; (F) Eszopiclone. Ligands were docked using Autodock Vina with the protocol developed elsewhere [144]. The structural domains I, II and III of Mpro are colored in light blue, orange and green, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The peptidomimetic α-ketoamides represent a class of prototypical inhibitors of Mpro. Molecular docking and molecular dynamics simulations were used to characterize the interaction of an α-ketoamide analogue with the active site of the SARS-CoV-2 Mpro. ùThe predicted Glide score and molecular dynamics indicated clearly that α-ketoamide analogues bind the Mpro more tightly than the amoxicillin used as control [128]. One of them, synthesized and defined as 11r, turned out to be particularly effective on MERS-CoV Mpro (K D ~ 400 pM) and moderately effective on SARS-CoV Mpro (K D ~ 2 µM) [129], resulting somewhat less powerful on SARS-CoV-2 Mpro (K D ~ 3 µM) [111].
The inhibitory power was then significantly increased (K D ~ 0.7 µM, see [111]) by substituting the P2 cyclohexyl moiety with a smaller cyclopropyl ring, suggesting that the S2 binding pocket is less flexible than originally thought. This was partially confirmed by a computational study, which, though observing a relevant flexibility of several domains of Mpro, detected a particular rigidity of its active site [130].
An interesting investigation identified a series of novel peptidomimetic aldehydes. These new compounds were designed so as to maintain the aldehyde as warhead in P1′ and holding the S1′ pocket of the Mpro active site. The most promising inhibitors, defined as 11a and 11b, exhibited anti-SARS-CoV-2 Mpro activity (IC50 = 0.053 μM and IC50 = 0.040 μM, respectively), being also effective on the anti-SARS-CoV-2 infection activity in cell culture (EC50 = 0.53 μM and EC50 = 0.72 μM, respectively). Both compounds showed also good results for a preliminary pharmacokinetic evaluation [125].
Natural products as Mpro inhibitors
Phytochemicals (i.e., molecules derived from plants and often adopted for food or traditional medicine purposes) are emerging in the very recent literature as an alternative source of investigation for the inhibition of SARS-CoV-2 Mpro. Hereafter, a series of in silico screening on these compounds is reported [131], [132], [133], [134], [135], [136], [137], [138], [139], [140].
A first interesting approach was carried out by screening an impressive number (more than 606 million) of compounds. Shape screening, molecular descriptors relevant for pharmacokinetics and complex stability (estimated by molecular dynamics simulations) were used to significantly reduce the number of molecules to be studied. A final list of 9 compounds was selected and the natural compounds (-)-taxifolin from plant of Pinaceae family and rhamnetin from plant of Myrtaceae family were identified as best binders and thus potential inhibitors of SARS-CoV-2 MPro [131].
Alternatively, a great number of compounds (more than 250), derived from Curcuma longa L. were tested and two of them, namely (a) the C1 (1E,6E)-1,2,6,7-tetrahydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione) and (b) C2 (4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxyphenyl)hepta-4,6-dien-3-one) revealed a better binding score (−9.08 and −8.07 kcal/mol, respectively) with respect to the control shikonin and lopinavir (around −5.4 kcal/mol) [132].
Bonducellpin D from Caesalpinia bonduc (Fig. 7B) showed high binding affinity (−9.28 kcal/mol) because its interaction is stabilized by four hydrogen bonds with Glu166 and Thr190 together with hydrophobic interactions established with eight residues. Moreover, Bonducellpin D exhibited a broad-spectrum inhibitory potential against SARS-CoV Mpro and MERS-CoV Mpro [133].
Three natural metabolites (like ursolic acid, carvacrol and oleanolic acid) passed ADME (i.e., Absorption, Distribution, Metabolism, and Excretion) property as well as Lipinski’s rule of five and were candidates as potential inhibitors of MPro [135].
Withanone, an active constituent from Withania somnifera, and the natural phenolic caffeic acid phenethyl ester component of propolis interact with the highly conserved residues of the substrate-binding pocket of SARS-CoV-2 Mpro, and their binding free energy were estimated comparable with that of the N3 protease inhibitor [135].
Three bioactive molecules from Camellia sinensis (i.e., oolonghomobisflavan-A, theasinensin-D, and theaflavin-3-O-gallate) were selected after docking and molecular dynamics simulation approach on SARS-CoV-2 Mpro, and they were compared with antiviral drugs. The oolong tea (blue tea)-derived molecule oolonghomobisflavan-A showed the best score and a higher number of hydrogen bonds network with respect to all tested compounds [136].
Metabolites and molecules from Indian spices, present in PubChem and Zinc databases, were also analyzed by bioinformatics approach, virtual screening tools and molecular dynamics. The best three molecules were a) carnosol from Rosmarinus officinalis, that exhibited hydrogen bonds interactions with residues present on the active site of SARS-CoV-2 MPro and also the highest binding affinity (−8.2 kcal/mol), b) rosmanol from Rosmarinus officinalis, (−7.99 Kcal/mol), and c) the triterpene glucoside arjunglucoside-I from Terminalia arjuna (−7.88 kcal/mol) [137].
An endemic plant Andrographis paniculate, adopted in traditional medicine, provides a potential inhibitor of Mpro, that is the andrographolide, which displays good solubility, target accuracy and obeys the Lipinski’s rule of five [138].
Further, three other natural compounds could be of interest, namely (i) hispidin from Pteris ensiformis, (ii) lepidine E, an alkaloid from the seeds of Lepidium sativum, and (iii) folic acid, which all bind tightly the enzyme forming hydrogen bonds with the residues of active site [139].
An additional source of potential SARS-CoV-2 Mpro inhibitors seems represented by natural marine products. For example, among five tested marine natural products, the cytotoxic molecule fostularin 3 exhibited hydrogen and hydrophobic interactions with residues in the active site of Mpro [140]. Additional promising inhibitors were phlorotannins from Sargassum spinuligerum, pseudotheonamides from sponge Theonella swinhoei, and also flavonoids. In particular, heptafuhalol A (Fig. 7C) showed the lowest docking energy (−14.60 kcal/mol), associated to a network of hydrogen bonds with the acceptor residues of Thr24, Ser46, Asn142, Glu166, Pro168 and hydrophobic interactions with Met49, Met65, Leu141, and Pro168. After the molecular dynamic simulation and re-docking protocol the His41 residue, belonging to the catalytic dyad, is shown to establish a hydrogen bond with the hydroxyl residue of the ligand [141].
Repurposed drugs as potential Mpro inhibitors
An effective alternative to non-specific Mpro natural inhibitors is provided by repurposed compounds, originally developed for other diseases or for protein inhibition in other human pathogens.
Virtual screening
In an interesting study two libraries of drugs were docked against Mpro using three of the most common docking programs. Only the molecules with high consensus among the different algorithms were considered new promising SARS-CoV-2 Mpro inhibitors. The predicted candidates were: perampanel, carprofen, celecoxib, alprazolam, trovafloxacin, sarafloxacin and ethyl biscoumacetate [142].
Alternatively, a virtual screening of FDA-approved drugs was performed against the SARS-CoV-2 Mpro, giving the glecaprevir and maraviroc as the best inhibitors [143]. Further, also selected anti-HIV drugs (e.g., saquinavir, ritonavir, lopinavir, and others) and anti-HCV drugs (e.g., simeprevir, faldaprevir, and asunaprevir) were docked against SARS-CoV-2 Mpro. Indeed, simeprevir (Fig. 7D), an approved HCV NS3/4A protease inhibitor, seems to be a promising lead compound, fitting quite well (-10.0 kcal/mol) in two hydrophobic pockets flanking the catalytic dyad dyad His41-Cys145 and inducing an opening of the substrate binding pocket, which weakens significantly the compactness of the active site [144].
Moreover, in addition to the aforementioned anti-HIV and anti-HCV drugs, a different study showed that best results were obtained with other compounds, like delavirdine, cefuroxime axetil, oseltamivir, and prevacid [145]. Furthermore, a study found that the anti-protozoal emetine and hespedin seem to bind nearby the catalytic residues His41 and Cys145. The molecules were surrounded by other residues identified as Met49, Gly143, His163, His164, Glu166, Pro168, and Gln189 [146].
Not only anti-HCV and anti-HIV drugs but also the anti-influenza triazavirin was docked against SARS-CoV-2 Mpro and tested clinically in China for COVID-19 treatment [147]. Viomycin, a nonribosomal peptide with antibiotic properties and administered for the treatment of Mycobacterium tuberculosis infection, was docked for Mpro embedding deeply inside the binding pocket and showing a higher number of hydrogen bonds with SARS-CoV-2 Mpro than N3 inhibitor [148]. A completely different category of drugs, like statins, have been docked with MPro; in particular pitavastatin, rosuvastatin, lovastatin, and fluvastatin may be novel molecules with inhibitory properties. In fact, pitavastatin (Fig. 7E) has a binding energy greater than that of protease inhibitors nelfinavir and lopinavir, reported in the same study [149]. Potential inhibitors of SARS-CoV-2 Mpro were also identified by a structure-guided virtual screening approach using inhibitor N3 as a starting reference. Interestingly, among the selected molecules, leupeptin, pepstatin A, birinapant, lypressin, and octreotide turned out to be remarkable potential inhibitors. All these molecules have applications spanning from anticancer therapy to widespread inhibition of different types of proteases [150]. A structure-based virtual screening on SARS-CoV-2 Mpro was also performed using the ChEMBL database and thousands of other compounds as reference. The first result discussed was a hit of 64 drugs classified into antibacterial, antidiabetic, anti-inflammatory, cardiovascular, gastrointestinal, anti-HIV, and neuropsychiatric drugs. Among them, two potential anti-SARS-CoV-2 drugs were obtained from autodock vina docking simulations, namely a) curcumin (−7.3 kcal/mol), b) sepimostat (−7.9 kcal/mol), even though the best score was found for the eszopiclone (−10.0 kcal/mol) (Fig. 7F) a drug used for the treatment of insomnia [151].
Further, the antibiotic talampicillin and the anti-psychotic lurasidone have been identified by virtual screening as potential drugs worth being tested against SARS-CoV-2 MPro [152].
An interesting molecular docking study compared the binding energy score of most promising molecules against SARS-CoV-2 MPro. It showed that O6K molecule had a binding score similar to N3 (−7.4 kcal/mol and −7.1 kcal/mol, respectively). Remdesivir and its metabolite (GS-441524) turned out to be slightly less effective (−7.0 and − 6.4 kcal/mol, respectively), while the entecavir, which has a structure similar to that of GS-441524, displayed also a closely similar score of −6.4 kcal/mol [153]. Notably, remdesivir, GS-441524 and entecavir might be multi-target potential inhibitors for both RNA-dependent RNA-polymerase (RdRp) and Mpro. In the same work, umifenovir, together with montelukast, a drug for the treatment of allergies and the prevention of asthma attacks, showed moderate binding score to Mpro (−6.5 kcal/mol and − 6.5 kcal/mol, respectively) while a lower score was found for chloroquine and hydroxychloroquine with −5.0 kcal/mol and −5.9 kcal/mol, respectively [153].
Experimental testing
An important set of promising molecules with inhibitory effect on Mpro were also tested against the purified enzyme, different cell lines, as well as by co-crystallization with Mpro.
Baicalin and baicalein, two compounds from Scutellaria baicalensis, were considered as novel non-peptidomimetic inhibitors of Mpro, displaying also an antiviral activity in SARS-CoV-2 infected cells. Baicalin showed an IC50 of 6.41 µM against the Mpro, while baicalein shows an IC50 of 0.94 µM; K d of baicalin and baicalein binding to Mpro were 11.50 and 4.03 µM, respectively [154]. The crystal structure revealed that baicalein binds Mpro in a core region of the substrate-binding site between domains I and II also interacting with the the His41-Cys145 dyad, S1/S2 sub-sites and the oxyanion loop.
Furthermore, Vero E6 cells were infected with SARS-CoV-2 in the presence of different concentrations of baicalin or baicalein. Baicalin and baicalein showed a dose-dependent inhibition on the replication of SARS-CoV-2 with an EC50 of 27.87 and 2.94 µM, respectively [154]. Therefore, the cell-based antiviral activity of baicalin or baicalein is close to that of chloroquine (EC50 = 2.71 µM) and hydroxychloquine (EC50 = 4.51 µM) [155] (see Table 2 ).
Table 2
Cell-based tested inhibitors of SARS-Cov-2 Mpro and their applications.
| Inhibitor | EC50 (µM) | Application | Refs. |
|---|---|---|---|
| Baicalin | 27.87 | Respiratory tract infection | [154] |
| Baicalein | 2.94 | Respiratory tract infection | [154] |
| Chloroquine | 2.71/5.47 | Malaria | [155], [162] |
| Hydroxychloroquine | 0.7/4.51 | Malaria | [162], [155] |
| Remdesivir | 23.15 | Ebolavirus | [161] |
| Boceprevir | 1.31 | HCV | [156] |
| Gly-Cys-376 | 0.91 | Picornavirus | [157]. |
| calpain inhibitor II | 2.07 | Ischemia | [156] |
| calpain inhibitor XII | 0.49 | Ischemia | [156] |
| Ebselen | 4.67 | Ischemia and inflammatory | [124] |
| N3 | 16.77 | SARS and MERS | [124] |
| Cinanserin | 20.61 | SARS | [124] |
| Lopinavir | 26.63 | HIV | [161] |
| Homorringtonine | 2.55 | Chronic myeloid leukemia | [161] |
| Emetine hydrochloride | 0.46 | Amoebiasis | [161] |
A second study displayed a variety of different drugs all tested against purified SARS-CoV-2 Mpro. The groups considered were proteasome inhibitors, HIV protease inhibitors, γ-secretase inhibitors, HCV NS3-4A protease inhibitors, DPP-4 inhibitors, miscellaneous serine protease inhibitors, cathepsin and calpain protease inhibitors, miscellaneous cysteine protease inhibitors, matrix metalloprotease inhibitors, and miscellaneous protease inhibitors [156].
Boceprevir, an anti-HCV drug, inhibited the enzymatic activity of Mpro with IC50 of 4.13 μM, and an EC50 of 1.31 μM against the SARS-CoV-2 virus in the primary viral cytopathic effect (CPE) assay. An interesting result was obtained with the protease inhibitor Gly-Cys-376 (a broad-spectrum inhibitor targeting Mpro in the picornavirus-like cluster), that showed promising antiviral activity with an EC50 = 3.37 μM as well as an enzymatic inhibition with a value of IC50 = 0.03 μM [156].
Interestingly, in another recent work, Gly-Cys-376 showed a promising high affinity (IC50 = 26.4 nM and EC50 of 0.91 μM) to SARS-Cov2 Mpro [157]. While ebselen and N3 displayed inhibition against SARS-CoV-2 with EC50 values of 4.67 μM and 16.77 μM, respectively [124].
Further, calpain inhibitors II and XII inhibited SARS-CoV-2 in the CPE assay with EC50 values of 2.07 and 0.49 μM, respectively, as well as the purified Mpro showing IC 50 = 0.97 μM and IC 50 = 0.45 μM, respectively. The common feature that confers inhibitory capacity of these promising drugs is the structural core of α-ketoamide (i.e., the warhead). The identified compounds were more powerful and selective than the reported SARS-CoV-2 Mpro inhibitors ebselen, N3, and 13b [156]. Previously characterized molecules, such as organo-selenium compound ebselen, disulfiram, tideglusib, and carmofur exhibited EC50 values of 0.67 μM, 9.35 μM, 1.55 μM, and 1.82 μM respectively [124].
Dipyridamole binds Mpro through hydrogen bonds and hydrophobic interactions, showing an inhibitory effect (IC50 of 0.55 μM) better than other drugs tested, such as the disulfiram (4.67 μM), which was taken as positive control for the bioassay [158]. Furthermore, dipyridamole and montelukast sodium showed a global inhibitory function on NF-kB signalling and inflammatory responses during viral infection [158].
Cinanserin was considered an inhibitor of SARS-CoV (IC 50 = 4.92 μM) [160], but the inhibition of the purified SARS-CoV-2 Mpro was not high (IC 50 = 125 μM), so as only moderate inhibition was observed against SARS-CoV-2 with an EC 50 of 20.61 μM from qRT-PCR analysis [124]. The cmpd-26, an analogue of cinanserin, showed an IC50 of 1.06 μM for SARS-CoV Mpro [159].
A pool of compounds, considered inhibitors of coronavirus replication in a clinical trial study, were validated for in vitro assay against SARS-CoV-2 virus in Vero E6 cells; they gave the following EC50: 23.15 μM (remdesivir), 26.63 μM (lopinavir), 2.55 μM (homorringtonine), and 0.46 μM (emetine hydrochloride), while ribavirin and favipiravir showed no inhibition at 100 μM. Only the synergistic effect of remdesivir at 6.25 μM with emetine at 0.195 μM achieved more than 60% inhibition in viral yield. The conclusion is that a “cocktail” of antiviral drugs may reduce compound concentrations and increase inhibition of viral replication [161].
Finally, chloroquine and hydroxychloroquine both inhibit SARS-CoV-2 in cell assays (EC50 = 5.47 μM vs 0.7 μM in the Vero cell infection model, respectively) [162], though displaying suboptimal affinity scores of − 5.0 kcal/mol and − 5.9 kcal/mol, respectively [153].
PLpro inhibitors
The lack of information pertaining PLpro, as compared with Mpro, in particular its mechanism of action and involvement in viral replication, does not rule out the possibility that it represents an excellent candidate for drug screening. In this respect, two substrates with natural and unnatural amino acids (i.e., Ac-hTyr-Dap-Gly-Gly-ACC and Ac-Abu(Bth)-Dap-Gly-Gly-ACC) were converted into inhibitors by exchanging the fluorescent tag (7-amino-4-carbamoylmethylcoumarin ACC) into the reactive group vinylmethyl ester. Both compounds exhibit high selectivity for SARS-PLpro variants and inhibit both SARS-CoV-PLpro and SARS-CoV-2-PLpro activities [122].
Two compounds, that is a) the GRL-0617 with an IC50 of 2.4 μM and b) compound 6 with an IC50 of 5.0 μM showed inhibition for SARS-CoV-2 PLpro [163]. GRL-0617 and compound 6 were selected and a plaque reduction assay was performed using Vero E6 cells and the SARS-CoV-2 USAWA1/2020; GRL-0617 and compound 6 exhibited EC50 values of 27.6 and 21.0 μM, respectively [163].
The biological evaluation (in Vero E6 cell and in HEK293 cell lines) of a second-generation series of SARS-CoV PLpro inhibitors (namely 3 k, 3e, 3j and 5c) has shown neither cytotoxicity nor off-target inhibitory activity. The 3 k compound exhibits the most potent PLpro inhibitory capacity (IC50 = 0.15 μM) and the highest antiviral effect in cell culture (EC50 = 5.4 μM) [164].
Clinical studies
In the last months, some of the drugs and compounds reported previously were employed in the treatment of COVID-19. Most of the drugs, identified by molecular docking or in vitro cell-based assay as potential inhibitors of SARS-CoV-2 Mpro, are actually potential therapeutic options, targeting different stages of SARS-CoV-2 “life cycle”. Therefore, only clinically oriented drugs are reported, such as chloroquine/hydroxychloroquine, Remdesivir, Umifenovir, Favipiravir and Lopinavir/ritonavir combination. The reason to use such drugs is that currently there are no drugs approved by FDA to specifically treat COVID-19.
The clinical observation clarifies that mortality is increased by co-morbidities like cardiovascular disease, hypertension, diabetes, chronic pulmonary disease, and cancer. The therapies with antiviral drugs may have important cardio-vascular side effects and toxicities, but the effect of short-term use of chloroquine/hydroxychloroquine, ribavirin, and lopinavir/ritonavir in patients without autoimmune diseases, hepatitis, HIV infection is not clear.
Remdesivir is an experimental drug for the treatment of Ebolavirus, so cardiovascular effects and toxicities are unknown, whereas chloroquine/hydroxychloroquine seem to have considerable cardiovascular effects, at least when used at high dosage [165]. Two other anti-HIV protease inhibitors, known to potentially inhibit SARS-CoV-2 Mpro [166], were employed in clinical trials, namely lopinavir and ritonavir, which were administered in adults hospitalized with severe COVID-19.
Lopinavir efficacy was modest and detected only in the early phase of SARS-CoV-2 infection, discouraging its use at the later stage of viral infection; further, lopinavir/ritonavir showed side-effects, such as nausea, diarrhoea and hepatotoxicity [167].
However, since infants and young children had relatively more severe illness than older children, a trial of hydroxychloroquine or lopinavir/ritonavir suggested their use in severe pneumonia and critically ill children [168].
The chloroquine and hydroxychloroquine, originally employed for malaria treatment, deserve a further clarification, since they should block the SARS-CoV-2 viral entry into cells by elevation of endosomal pH and by inhibition of the ACE2 terminal glycosylation [169], thus ultimately interfering with virus receptor binding.
Unfortunately, the clinical data and the effect of hydroxychloroquine in combination with azithromycin are mostly controversial about their efficacy in COVID-19 treatment. Hydroxychloroquine and chloroquine have certain limitations and toxicity, especially on the heart and eyes. Considering their ocular, cardiac and neuro toxicities, hydroxychloroquine and chloroquine should not be recommended as preventive drugs for the COVID-19 pandemia [170]. A larger, randomized, placebo trial with a prolonged follow-up will be required in the near future [171].
Remdesivir is a nucleotide analogue prodrug that inhibits RNA-dependent RNA-polymerase, but it is not a Mpro protease inhibitor and only in a computational study it was suggested that may be an inhibitor of both proteins [153]. However, a clinical study about the compassionate use of remdesivir for patients with severe COVID-19 revealed a clinical improvement in 36 of 53 patients (68%), even though a placebo-controlled trial will be required [172]. In a second study, the remdesivir was not associated with statistically significant clinical benefits in adult patients with severe COVID-19, but a larger study will be required to confirm the data [173].
In a non-randomized study of 67 patients, affected by COVID-19, a lower mortality rate was shown to occur in patients treated with umifenovir (arbidol hydrochloride), as compared with patients who did not receive the drug [171]. Moreover, in a small group of patients without invasive ventilation, an interesting study showed that oral arbidol plus lopinavir/ritonavir was associated with a significant negative conversion rate of coronavirus test in 7-day and 14-day, with respect to the lopinavir/ritonavir therapy group [174]. Umifenovir is not a protease inhibitor itself and inhibits membrane fusion of the viral envelope by targeting the interaction of the S protein with ACE2.
Favipiravir, an anti-influenza drug approved in Japan, interferes with viral replication and it is a potential inhibitor of RNA-dependent RNA-polymerase. A randomized study, conducted in a Chinese medical centre, compared umifenovir (200 mg3/day) and favipiravir (1600 mg2/first day followed by 600 mg*2/day) showing that, unlike umifenovir, favipiravir did not significantly improve the clinically recovery rate at Day 7 [171].
reference link : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7501082/
More information: Alice Douangamath et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease, Nature Communications (2020). DOI: 10.1038/s41467-020-18709-w



{kind=link}