











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Could something as simple as a certain type of sugar water be medicine for perforated bones, and even bone marrow cancer itself?
Inside our bodies are some jellyfish-like cells that actually eat away at our bones. Every year, they eat about ten percent of the bone mass in our body. Fortunately, other cells usually follow and build up new bone.
We undergo a kind of continuous remodeling and repair that enables most of us to traipse around with steel in our legs and arms.
In people with bone marrow cancer, the bone-eating cells run amok. They become too numerous and eat too much. The bone-building gang doesn’t have time to rebuild the bone mass, despite overtime and long shifts. Bone tissue gets gobbled up.
Many people with bone marrow cancer often end up with perforated bones, a condition that is very painful to live with. They sometimes experience collapsed vertebrae or suffer broken bones just by turning in bed.
For decades, scientists around the world have been scratching their heads and wondering what the cause could be. Various theories have been launched, but researchers have not reached a unified theory on what the main cause is.
Too many unusable antibodies produced
Bone marrow cancer remains an incurable disease so far. The available treatment can prolong life, but not cure the disease.
Now Standal and her research group at CEMIR at NTNU have discovered a piece of the puzzle that looks very promising.
They have come to the conclusion that the cause of the bone destruction is too little sugar. We’re not talking about the sugar we eat in our cakes and biscuits, but sugar that resides on a substance that is important for the immune system.
To get to the bottom of how sugar is related to bone loss, we need to get into the bone marrow. This is the soft cavity that inside all our bones.
Within the bones are plasma cells. When bacteria or viruses enter the body, the plasma cells begin their job of getting rid of the invaders. Antibodies are produced which are sent via the blood, ready to do battle.
So far so good, but in people with bone marrow cancer, far too much of one type of antibody is produced. It’s going amok here, too. The antibody that the cancer makes is also completely useless.
It doesn’t knock out either the cold or the flu but just takes up too much space and displaces other types of antibodies.
Search for an answer took five years
“I thought simply. If people with bone marrow cancer have too much of the antibody and too many bone-eating cells, then they must be connected,” Standal says.
The search for an answer gobbled a lot of her working hours for almost five years. The hard work was fortunately not in vain, and has led to a completely new and fundamental understanding.
The finding has now been published in the highest ranked blood disease journal in in the world.
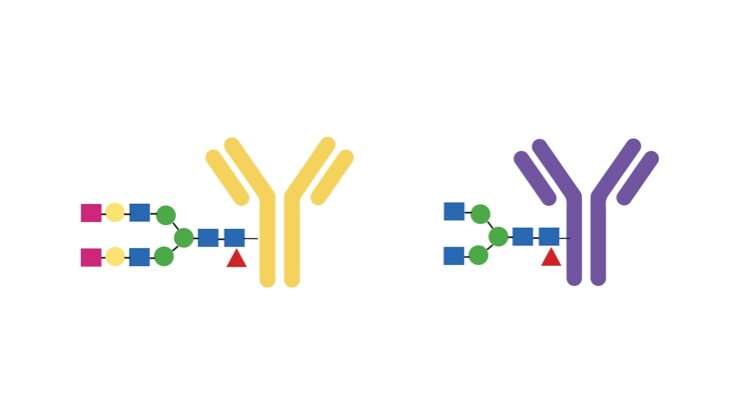
This is how Standal arrived at the answer:
The vast majority of patients with bone marrow cancer develop perforated bones, but not all. Standal asked nicely, and received samples from patients with bone loss. She also took samples from patients without this kind of bone loss.
The researchers extracted antibodies from the samples and cultured bone-eating cells in the laboratory.
- When Standal placed the bone-eating cells into the antibody of the patients with bone perforations, she discovered that the number of bone-eating cells increased.
- When she put the bone-eating cells into the antibody of the patients without bone perforations, she discovered that the number of bone-eating cells did not increase.
Removed and added sugar
“Why that was the case became the next interesting thing to figure out,” Standal says.
The antibody carries a type of sugar that “decorates” it, in a way. The sugar has an effect on how the antibody works. Standal found her way to Manfred Wuhrer at the Center for Proteomics and Metabolomics of the Leiden University Medical Center in the Netherlands. He is a specialist in this type of sugar, and Standal sent the samples to him.
He found that individuals with bone loss were missing two sugar molecules at the end of a long chain inside the antibody.
“There was too little sugar,” says Standal.
But this answer wasn’t sufficient, either.
Although a difference was detected between the two groups, the researchers could not confirm that the missing sugar molecules were the reason patients developed more bone-eating cells. Several further experiments had to be conducted.
The research team went to the lab and put more sugar on the antibody. This did not lead to more bone-eating cells. Standal also did the opposite, removing sugar from the antibody. This did lead to more bone-eating cells.
Fed mice sugar water
The researchers then had sufficient test results to show that too little sugar can be decisive for the number of bone-eating cells. But this is not enough in medical research—at least not if the goal is to use the knowledge to make medicine for humans.
The next step involved animal experiments with mice that have bone marrow cancer. The mice were divided into two groups and were given two different types of sugar water. In theory, one type of sugar water would lead to more sugar on the antibody.
“The theory actually worked. The mice that received this type of sugar water had smaller perforations in their bone tissue. They also developed less cancer,” says Standal.
Now she has to carry out more animal experiments to move forward on the path towards a treatment that can give patients with bone marrow cancer a better life.
“I think it might be realistic to try this on a small group of patients in four to five years,” says Standal.
Life forms have a dynamic nature. Within organisms, energy is constantly produced and consumed, and chemicals, including protein, fat, and sugar, undergo constant synthesis and degradation. A stable chemical and energy intake, either in the form of food absorbed in the digestive system or sunshine converted in the leaves, is indispensable for life to survive and multiply. For a long time, people believed that energy or chemicals from the outside environment are the sole source to support synthesis in vivo and maintain homeostasis.
Later, we recognized that, under adverse environments, such as starvation, intracellular recycling of chemicals, including protein, fat, and minerals, became a backup solution to maintain the minimal amount of synthesis and energy production needed for survival.1
Furthermore, it has now been acknowledged that even under normal physiological conditions, the substrates for most intracellular synthetic processes in our body are mainly derived from the degradation, reformation, and reuse of contents that are already present. Such recycling characteristics become another intrinsic definition of life and are mainly achieved through a biological process named autophagy.2
Generally, autophagy is a highly conserved intracellular catabolic process during evolution, in which cytoplasmic components are degraded for nutrient and/or energy generation. At the very beginning, the physiological function of autophagy was limitedly recognized as merely a way of transporting intracellular components to the lysosome.
Ever since the identification of autophagy-related genes and the molecules involved in membrane dynamics during autophagy, however, significant progress has occurred regarding the broad participation of autophagy in almost all biological processes.
Autophagy has been identified as a critical player in both physiological processes and the onset and progression of numerous pathological conditions related to metabolic dysregulation,3 including cancer,4 neurodegenerative disorders,5 aging,6 and bone-related diseases.7
Under physiological conditions, autophagy is responsible for the removal of damaged or excessive organelles, whereas under pathological conditions, autophagy helps in the redistribution of intracellular nutrients to meet the substance and energy requirement for survival. Autophagy controls the energy and chemical homeostasis of each single cell and various tissue types, including bone.8
In mammals, bone assumes multiple important functions, providing protection to vital organs, attachments for skeletal muscles, niche sites for blood cell synthesis, a form of storage for mineral ions, and secretion organs of hormones.9 To fulfill the above-mentioned roles, our skeletal system undergoes a constant remodeling cycle.10 MSC (mesenchymal stem cell)-derived osteoblasts lining the surface of bone synthesize and secrete bony matrix.11
The matrix-embedded osteoblasts further differentiate into long-lasting osteocytes, and the latter forms a mechanosensing network within the bone.12 At the same time, multinucleated osteoclasts derived from hematopoietic stem cells constantly degrade and resorb their surrounding bone matrix. Normally, a dynamic balance between the formation and degradation of the bone is constantly coordinated.13 In this way, the mass, structure, and functions of bone tissue are unsurprisingly sensitive to either intrinsic or extrinsic stimuli.
When the equilibrium between bone formation and bone degradation is disturbed, pathological conditions occur. Excessive bone-formation activity leads to overmineralization and excessive mass of bone, which is called osteopetrosis, also known as marble bone disease.14 However, when an imbalance toward bone degradation predominates, increased bone loss leads to reduced mass and undermined structure of bone, which is frequently called osteoporosis.15
Osteoporosis is a systemic bone degenerative disease characterized by progressive loss of bone mass and significant degradation of bone mechanical properties, which subsequently leads to bone fragility and susceptibility to fractures. This phenomenon is usually correlated with the progress of aging and significantly worsens the quality of life and longevity of the elderly population.16,17
Considering the “recycling” property of autophagy and the dynamic synthesis and degradation processes within bone, it is not surprising to find that autophagy is highly involved in the metabolism of bony tissue. All three types of bone cells, osteoblasts, osteocytes, and osteoclasts, demonstrate a basal level of autophagic activity. Multiple components of the autophagic pathway contribute to mediating the survival and functioning of osteoblasts, osteocytes, and osteoclasts.18–20
Increasing evidence suggests that an appropriate level of autophagy enables osteoblasts, osteocytes, and osteoclasts to survive hypoxic, nutrition-deficient, or even hypertonic environments. In addition to survival, the level of autophagic activities is associated with pre-osteoblast differentiation, osteoblast-osteocyte transition, and the genesis and functioning of osteoclasts.18,21,22
In addition to the correlation between autophagy and bone physiology, recent evidence suggests that autophagy plays a fundamental role in the onset and progression of pathological osteoporosis.7,22,23 The relationship between the autophagy pathway and osteoporosis was highlighted in a genome-wide association study of human wrist bone mineral density, where significant correlations between multiple autophagy-regulatory genes and bone mineral density were identified.24
In addition, selective modulation of autophagy-related genes in bone cells is sufficient to recapitulate the osteoporotic state in animal models.18,25 At the same time, modulation of autophagic activities has potential therapeutic value for the prevention and treatment of osteoporosis.26,27
This review summarizes the up-to-date research findings about the autophagic process and its role in skeletal homeostasis and the onset of osteoporosis, and discusses both the potential and challenges in the therapeutic application of autophagy modulators.
Autophagy: self-eating for survival
Autophagy, which originates from the Greek roots autos- (self) and phagein- (eat), is a lysosomal pathway responsible for the recycling of unnecessary cell organs and excessive nutrients and the elimination of metabolic wastes and intracellular pathogens.26–28 Such an intracellular “self-eating” process plays a critical role in maintaining the survival of multiple cell lineages.29
In mammals, three types of autophagy with distinct morphological features and different regulatory mechanisms have been described: chaperone-mediated autophagy, microautophagy, and macroautophagy. In chaperone-mediated autophagy, cytoplasmic proteins are not sequestered and are delivered to the lysosome by chaperone proteins rather than membranous structures.30 Chaperones match with proteins containing a specific pentapeptide motif, and then, these substrates are unfolded and translocated individually and directly across the membrane of lysosomes.31
In microautophagy, the lysosome directly captures a small amount of nearby cytoplasm by forming invaginations or protrusions on its membrane, requiring little assistance from organelles outside the lysosome.32–34 In macroautophagy, the capture and delivery of intracellular substances is symbolized by the formation of autophagosomes. The autophagosomes are constituted by newly formed bilayer membranes, and can enclose damaged organelles, intracellular pathogens, and protein aggregates to achieve the sequestration process.35
Autophagosomes are then incorporated by lysosomes to finish the delivery and digest the contents (Fig. (Fig.1).1). Compared with microautophagy, autophagosomes can capture cytoplasm far away from the lysosome. Macroautophagy is regulated by a group of evolutionarily conserved genes named Atg (autophagy-related genes). The Atg genes have diverse functions, including the transportation of both intracellular and extracellular cargos and coordination of intracellular communication with all kinds of signaling pathways.
The Atgs include approximately 20 members. During the initiation and maturation of autophagosomes, Atgs are actively involved in the formation of double-membrane vesicles and the delivery of cargos in autophagosomes to lysosomes.36 Meanwhile, Atgs may interact with signaling pathways other than autophagic ones. For example, Atg7 is downstream of FGF signaling in the regulation of endochondral bone formation and long bone growth.37

Three types of autophagy. Schematic illustrations of (a) macroautophagy, (b) chaperone-mediated autophagy, and (c) microautophagy
Among the three types of autophagy, macroautophagy has the strongest connection with cell biology, physiology, and disease, and will hereinafter be referred to as “autophagy” in this review.
A highly organized degradation program
Autophagy is a highly conserved cellular process during evolution.2 From yeast to vertebrates, autophagy works in concert with the UPS (ubiquitin–proteasome system) to maintain cellular homeostasis.38 Closer examination defines the autophagic process into four major stages: initiation/nucleation, elongation, degradation, and termination (Fig. (Fig.22).32,35

Major stages in the autophagic process. Schematic illustrations of major stages in the autophagic process: initiation and nucleation, elongation, closure and maturation, fusion and degradation
Autophagy starts with activation of the ULK1 complex, which is composed of ULK1, ATG13, ATG101, and FIP200. The ULK1 complex originally associates with the mammalian target of rapamycin complex 1 (mTORC1) complex. At the initiation of autophagy, ULK1 is dephosphorylated, and the ULK1 complex dissociates from mTORC1.39
The activated ULK1 complex recruits another multiprotein complex, known as the class III phosphatidylinositol 3-kinase (PI3K) complex, to the site of autophagy initiation. The PI3K complex is composed of beclin-1, Vps15, Vps34, Ambra1, UVRAG, and more.28,40 Ambra1 interacts with TRAF6 and leads to self-association and stabilization of these complexes. In this process, a membrane fragment usually known as a phagophore is formed.41
In the next step, ATG proteins participate in the elongation of the phagophore. The ATG proteins aggregate and form a ubiquitin-like conjugation system, ATG12–ATG5–ATG16L, which facilitates the assembly of LC3 (microtubule-associated protein 1A/1B-light chain 3) with PE (phospholipid phosphatidylethanolamine). LC3-PE, which is also called LC3-II, then incorporates into the phagophore membrane and contributes to the elongation and closure of the autophagosome.32,42
Autophagosomes mature by fusion with intracellular endocytic components, including endosomes and lysosomes,43 turning the environment inside the autophagosome acid. Proteins involved in vesicular transport, such as dynein, and membrane fusion, including Rab7, SNARES, and ESCRT, facilitate the maturation of autophagosomes.44
Some proteins on the surface of autophagosomes, including p62, optineurin, NDP52, NBR1, and Alfy,45,46, are responsible for the sequestration of degradation targets. During the degradation stage, entrapped intracellular macromolecules are broken down into amino acids, lipids, nucleotides, and energy for the purpose of future intra- and extracellular processes.47
Termination of autophagy is achieved through a negative feedback mechanism. Nutrients produced in autophagosomes reactivate the mTOR (mammalian target of rapamycin) pathway, and the latter generates proto-lysosomal tubules or vesicles. These tubules and vesicles extrude from the autolysosomes and eventually mature into lysosomes again. Such a termination process serves as the closing stage of the autophagic machinery and has been validated in various species.48,49
Critical molecules in the above-described autophagic process have been employed for the assessment of autophagy flow. For example, Beclin-1 is fundamental for the formation of PI3K complexes and, therefore, has been commonly used as a marker of autophagic initiation.48 LC3-II found within the autophagosome membrane has been widely used as a specific autophagosome marker.32,49 Analyses of the combined expression of proteins p62 and LC3-II are commonly used to assess autophagic flow.50,51
In addition to degrading intracellular contents, autophagy can target extracellular cargo. Several core ATG proteins are involved in the phagocytosis of unwanted extracellular components. During such ATG-assisted phagocytosis, extracellular targets, such as pathogens and apoptotic cells, are engulfed by single-layered vacuoles and then labeled by LC3, which delivers the contents to lysosomes for degradation.52,53
A target-specific digestion process
For a long time, autophagy has been recognized as being nonselective for its degradation substrates.50,51 The simultaneous observation of multiple intracellular components in double-membrane vesicles has been employed as a standard for the identification of autophagy.
While this is often true when autophagy is induced in stressed conditions such as starvation, recent evidence suggests that autophagy required during the maintenance of cell homeostasis could be highly specific.51,54–57 Actually, autophagy can be extremely specific in choosing the cargo for autophagosomes. An intricate system is in charge of barcoding and selectively sequestering the substrates for autophagy.
This process is termed selective autophagy and is more critical during diseases. Substrates commonly sequestered and digested during selective autophagy may include ubiquitinated proteins, peroxisomes, and mitochondria (Fig. (Fig.33).58–60

Non-selective and selective autophagy. Schematic illustrations of starvation-induced nonselective autophagy and target-specific selective autophagy
The best demonstration of protein-specific autophagic degradation is the ubiquitin-binding protein SQSTM1 (sequestosome 1), also called p62, on the autophagosome surface. P62 can capture ubiquitinated proteins and binds to the membrane component LC3-II.61
While delivering the target substrates to the inside of the autophagosome, p62 itself is also internalized and degraded. P62 is considered one of the major digestive substrates for autophagosomes, and therefore, increased expression of p62 usually indicates a decline in the autophagic process. NBR1 autophagy cargo receptor and OPTN (optineurin) are other receptors that specifically deliver ubiquitinated proteins or pathogens to autophagosomes.62–64
Ubiquitination-involved autophagy is most active in the clearance of bacteria. When the pathogens are specifically engulfed and digested during autophagy, the process is named xenophagy.62,65
When peroxisomes are selectively degraded during autophagy, the process is termed pexophagy. LC3-II on the surface of autophagosomes could specifically bind to peroxisomal biogenesis factor 14 (PEX14), a component of the peroxisomal translocon complex. In this way, peroxisomes are specifically recognized by autophagosomes during starvation.66
Actually, in certain organs, autophagy could accomplish up to 70%–80% of peroxisome turnover under normal growth conditions.67 Considering the widespread influence of peroxisomes on metabolism and the negative consequences of peroxisomal dysfunction in health, specific pexophagy must be a critical player in maintaining organism homeostasis.58
Another selective target for autophagy is the mitochondria, and the specific autophagic degradation process is termed mitophagy. For intact mitochondria, LC3 and gamma-aminobutyric acid receptor-associated protein (GABARAP) on expanding phagophores could specifically bind to a complex on the outer membrane of the mitochondria called BNIP3L/NIX and subsequently turned the following maturation and degradation stages into mitophagy.68
For damaged mitochondria, this selective capture process is slightly altered. When a mitochondrion loses its integrity, the kinase PINK1 accumulates on its outer membrane and specifically binds to the cytosolic E3 ubiquitin ligase PARK2/Parkin. The latter then ubiquitinates mitochondrial substrates and initiates mitophagy.68 Mitophagy is responsible for the routine turnover of mitochondria under normal conditions69 and is also important to the differentiation of certain cell lineages.70–72
For instance, the maturation of mammalian red blood cells requires the elimination of the mitochondria from immature cells, and this process is mainly achieved through mitophagy.72–74 Insufficient clearance of the damaged mitochondria has been associated with multiple pathological conditions. In autosomal recessive Parkinson’s disease, the genes encoding PINK1 and PARK2 are mutated,75,76 suggesting the critical role of mitophagy in maintaining cellular and organismal health.
In addition, autophagy could selectively target misfolded proteins that tend to aggregate. These inappropriate protein products are usually involved in pathological states, including neurodegenerative, skeletal, cardiac muscle, and liver diseases.
Selective autophagy demonstrates concurrent mechanisms in recognizing specific digestive substrates. These coexisting mechanisms cooperate with each other in a redundant manner to ensure efficient digestion of the unwanted material.
Physiological and pathological autophagy inducers
Autophagy is initiated with either physiological signals or pathological stimuli. At the physiological basal level, the autophagic process is constitutive at a low level in all cells, serving as a quality control mechanism to remove flawed organelles and proteins.77 The basal level of autophagic activity varies among different cell lineages and tissue types.78 Generally, such basal-level autophagy is more critical for highly or terminally differentiated, long-lived cells, such as neurons, myocytes, and osteocytes, in maintaining their homeostasis and functioning.79
A wide range of extracellular and intracellular stresses, including nutrient or energy starvation, hypoxia, disturbance in growth factor level, or pathogen invasion, induce an increased rate of autophagy to recycle cytoplasmic components into metabolites and biosynthetic processes or to eliminate pathogens, allowing for cell survival.80–82
The cAMP-dependent PKA (protein kinase A) pathway and mTOR pathway are involved in the initiation of autophagy induced by nutrient starvation. These two pathways sense levels of carbon and nitrogen.83 In conditions where nutrients are sufficient, PKA inhibits autophagy by inducing phosphorylation of LC3.84,85 A high level of amino acids upregulates RAG (RAS-related small GTPases), which subsequently activates MTORC1 and inhibits autophagy.86,87
While the PKA pathway and TOR pathway could both independently affect the Atg1/Atg13 protein kinase complex and its subsequent autophagic process,88 there might also be crosstalk between them. For example, the PKA and TOR pathways are both nutrient-sensitive, and their function overlaps in regulating cell proliferation.89 The regulation of ribosome formation from TOR is achieved partially via PKA signaling.90 Thus, these two pathways might coordinate in the fine-tuning of the level of autophagic activity.
In mammals, PKA can phosphorylate and activate MTORC1.91,92 PKA can also indirectly activate the MTORC1 complex through the inactivation of AMP-activated protein kinase (AMPK).93 In addition to being a substrate for PKA, AMPK is a major intracellular energy-sensing kinase. AMPK senses AMP/ATP levels and regulates a wide variety of cellular processes, including autophagy.94,95 At a low energy level, AMP binding activates the kinase activity of AMPK, and the latter phosphorylates and activates the TSC1/2 complex, which indirectly inhibits the function of MTORC1.96,97 At the same time, AMPK may directly downregulate the activity of MTORC1.98,99 Evidence also suggests that AMPK can phosphorylate and activate ULK1, which subsequently induces autophagy.100–103
Hypoxia and disturbance in growth factors also induce autophagy via the TOR pathway. Even with adequate nutrients and energy supply, hypoxia or growth factor fluctuation can inhibit MTOR1 and result in the induction of autophagy.95,104,105
In the case of endoplasmic reticulum (ER) stress, the increase in cytosolic Ca2+ concentration activates calcium/calmodulin-dependent protein kinase kinase 2/beta (CAMKK2/CaMKKb), and the latter subsequently activates AMPK and induces autophagy.106 At the same time, when unfolded proteins accumulate in the ER under stress conditions, unfolded protein response (UPR) signaling is triggered to induce autophagy. The ending point of ER stress-induced autophagy is still unclear. Both enhanced survival and increased autophagic apoptosis have been reported in the current literature.82,107
In addition to trafficking substrates to autophagosomes for degradation, ATG proteins are involved in the process of exocytosis. LC3 regulates this autophagic-independent exocytosis, which expels pathogens that either locate inside the autophagosomes or are labeled by LC3.52 In the case of virus invasion, ATG proteins are involved in the formation of membrane-associated replication units of virus or bacteria, including hepatitis C and Brucella abortus.108,109
Generally, autophagy contributes to cell homeostasis by specifically eliminating flawed or redundant organelles and boosting chemicals and energy recycling.98 Under most conditions, autophagy serves as a cytoprotective mechanism, but could potentially turn deleterious if it becomes uncontrolled. Autophagic dysfunction is thus associated with a wide variety of human pathological conditions, including skeletal system disorders and diseases.36,110
Autophagy regulates bone formation
The development, growth, and maintenance of the skeletal system are in dynamic balances and are highly sensitive to factors, including mechanical stimulus and hormone fluctuation.111 Mesenchymal cells in the bone marrow or lining the periosteum include a multipotent population that is capable of osteogenic, adipogenic, and chondrogenic differentiation. Mesenchymal-derived osteoblasts are responsible for the synthesis, secretion, and mineralization of the bony matrix and further differentiate into osteocytes when embedded in the mineralized matrix.112
Osteocytes are embedded in the bone matrix and form a cellular network that regulates skeletal remodeling.113 Hematopoietic stem cell-derived multinucleated osteoclasts execute bone resorption during remodeling. Osteoclasts secrete degradative enzymes to the bony surface to dissolve the minerals and digest the bony matrix and recycle the degraded contents via endocytosis.114 These three types of cells are tightly linked to orchestrate the homeostasis of the skeletal system (Fig. (Fig.4).4).
For example, the physiological stimulus for osteoblast-mediated new bone formation is strongly connected with the resorptive activities of osteoclasts during which growth factors such as TGF-β1 (transforming growth factor β1),115 IGFs (insulin-like growth factors),116 and bone morphogenetic proteins (BMPs) were released from the extracellular matrix of the bone.112,117,118 Autophagy is actively involved in each component of this cross-talking network.

Bone remodeling and skeletal system homeostasis. Osteoblasts, osteocytes, and osteoclasts are tightly linked to regulate bone remodeling and orchestrate the homeostasis of the skeletal system
Phenotypically, human genomewide screenings have illuminated the correlation between single-nucleotide polymorphisms of autophagy-related genes and status.119 The expression of autophagic pathway-regulatory genes demonstrated a direct influence on the variation in bone mineral density (BMD) in the distal portion of the radius.24,120
Among cells with a high secretion capacity, autophagy controls the spatial localization of signaling complexes critical to protein synthesis.121 Such intracellular spatial localization has been revealed in the activation of Wnt122 and NF-kB123 signaling via autophagic degradation of specific pathway components. In addition to protein trafficking, autophagy orchestrates diverse cellular pathways involved in cell viability, cell renewal, and innate immune response.
Since these pathways are critical to the differentiation of osteoblasts and osteoclasts, the importance of this autophagy-regulated spatial localization in regulating the anabolic and catabolic functions of bone cells is apparent. Since autophagy is an indispensable cell activity during embryogenesis, whole-body deletion of autophagy-related genes in animal models almost invariably causes death at birth.79 As a result, various cell-specific conditional knockout models have been established to explore the specific role of autophagy in osteoblasts, osteocytes, and osteoclasts.124
Autophagy fine-tunes osteoblast differentiation and functioning
Osteoblasts are the primary constructors of bone. These mesenchymal-derived cells deposit the bone matrix via constant synthesis and section activities. Both survival and physiological functioning are closely regulated by autophagy.
In the bone marrow, a bone-fat balance has been described in the differentiation of mesenchymal stem cells. Aging, estrogen deficiency, or a high-fat diet would push MSCs toward adipogenic differentiation and result in compromised bone density. Autophagy has been correlated with the stemness maintenance of mesenchymal progenitors.125
MSCs were shown to accumulate autophagosomes in the stem state and deliver them to lysosomes once differentiation is initiated, suggesting that autophagy-related metabolism is tightly associated with MSC differentiation.126 More specifically, the activation of autophagy has been correlated with the osteogenic differentiation of MSCs through AMPK signaling pathways.127
An appropriate autophagy level is a prerequisite for the maintenance of homeostasis and survival of osteoblasts. In vitro studies demonstrated a negative correlation between the level of autophagy and oxidative stress. Pharmacological downregulation of autophagy leads to increased oxidative stress in osteoblast-like cells, whereas upregulation of autophagy in these cells is correlated with reduced oxidative stress and decreased apoptosis.128
Knockdown of autophagy-essential genes increased the level of oxidative stress in osteoblasts.18 Additional data suggested that the damage caused by oxidative stress to osteoblasts could be relieved by early initiation of autophagy, which might be achieved through the ER stress pathway. Estrogen has demonstrated inhibitory effects on the serum deprivation-induced apoptosis of osteoblasts, and part of this protective effect is achieved by promoting autophagy via the ER-ERK-mTOR pathway.129
In addition, autophagy demonstrated protective effects on osteoblasts from various toxic stimuli. For example, a high level of autophagic flux reduced cell death of osteoblasts exposed to lead chloride,130 and upregulation of the level of autophagic flux aided in the survival of differentiated bone cells in a stressful environment.131
In addition to survival, autophagy is closely related to the differentiation and mineralization of osteoblasts. The most direct proof of the role of autophagy in mineralization is the identification of apatite crystals in autophagy vacuoles.18 Inhibition of autophagic flux also blocked the outward transportation of the minerals from osteoblasts. Gene knockout of Beclin-1 or Atg7 resulted in deficient mineralization in an in vitro setting.18
The in vivo role of autophagy in mineralization has also been reported. Targeted Atg5 deletion in osteoblasts led to a 50% reduction in the trabecular bone mass.132 Osteoblastspecific deletion of Fip200 (FAK family–interacting protein of 200 kDa) undermines the terminal differentiation of osteoblasts, inhibits bone formation, and consequently leads to an osteopenia phenotype.133
Moreover, osteoblast-specific conditional deletion of Atg7 led to decreased bone formation by triggering ER stress, and relief of ER stress by systemic delivery of phenylbutyric acid could restore the bone-formation balance.134 When Atg7 was deleted in the entire osteoblast lineage using Osterix-Cre transgenic mice, the number of both osteoblasts and osteoclasts decreased, and the osteocytes demonstrated decreased cellular projections and increased ER retention.135
Autophagy is also actively involved in signaling pathways that are of confirmed significance to osteogenesis. For example, insulin-like growth factor-I (IGF-I) stimulates the osteogenic differentiation of osteoblasts, the function of which is at least in part achieved via activation of AMPK and upregulation of autophagy.136
In addition, one of the pro-osteogenic cascades induced by bone morphogenetic protein-2 (BMP-2) involves the activation of the autophagy-related factor Atg7, which subsequently targets Wnt16 to activate metalloproteinase-13 and eventually osteoblastic differentiation.137
In addition to osteoblasts, chondrocytes are another critical cell population responsible for skeletal growth. Except for the craniofacial region, long bones form and grow via endochondral bone formation, during which chondrocytes undergo hypertrophy and active matrix secretion.138 In vitro studies revealed that the differentiation and mineralization of chondrocytes are positively correlated with the level of autophagy activity.139
During the postnatal growth of mice, autophagy is turned on in the growth-plate chondrocytes, and its level is closely correlated with the secretion of type II collagen, which is the major component of cartilage matrix. When Atg7 is deleted specifically in chondrocytes, the synthesized type II procollagen could not be transported out but was retained within the ER.
Furthermore, autophagy in growth plate chondrocytes was suppressed in Fgf18 and Fgfr4 deletion mice, resulting in osteopenia. This phenotype could be rescued by pharmacological activation of autophagy. These data suggest that autophagy serves as an effector for FGF signaling during endochondrial bone formation.37
Multiple pathways or growth factors with evident bone-regulating capacities demonstrate crosstalk with autophagic activities. BMPs are recognized as strong osteogenic growth factors, and members of the protein family have been successfully applied in clinical settings. BMPs directly bind to receptors on the surface of osteoblasts and activate the bone-formation process through the intracellular SMAD signaling system.
The paracrine function of the BMPs could be adjusted by the level of its extracellular antagonists, noggin, chordin, and sclerostin.140 The evidence from a study in pancreatitis cells suggested that BMPs may antagonize the dampening effect of noggin on microtubule-associated protein 1 light chain 3 (MAP1LC3)-II levels and subsequently increase the expression levels of Beclin-1 and lysosomal-associated membrane protein 2 (Lamp2). In this way, BMP ligands might be involved in the regulation of autophagy levels.
β-catenin-dependent canonical Wnt signaling is another osteogenic pathway that has been associated with autophagy. Wnt signaling is critical to the commitment of stem cells to the osteoblast lineage and the subsequent osteoblast differentiation. Wnt ligand triggers DVL (dishevelled segment polarity protein) recruitment to the plasma membrane by binding to the Frizzled receptor.
DVL promotes MAP1LC3-mediated autophagosome recruitment, ubiquitination, and degradation by binding to SQSTM1. In addition, under stress conditions, DVL becomes ubiquitylated and recognized by p62, and p62 in turn promotes DVL aggregation and degradation by LC3-mediated autophagy.122 In this way, the Wnt signaling pathway is negatively associated with autophagy. Moreover, since activation of the WNT-CTNNB1 signaling pathway is critical to the pathogenesis of osteoarthritis, activation of Wnt signaling might suppress autophagic activity and increase osteoblast or chondrocyte apoptosis.
At the same time, multiple autophagy-related proteins have been suggested to more directly influence the biology of osteoblasts. For example, knock-in mutation of the autophagy cargo receptor NBR1 resulted in enhanced osteoblast differentiation and bone formation.141 NBR can identify ubiquitinated proteins by its ubiquitin-like modifier activating enzyme domain and deliver them to autophagosomes by binding to MAP1LC3 by its LC3-interacting domain LIR.142 Aberration in these two domains of NBR1 leads to increased expression of SQSTM1 on the surface of autophagosomes and many other cytoprotective factors.
Two families of transcription factors with evident functions in autophagic activities are involved in the survival, differentiation, and function of osteoblasts. Members of the forkhead box O (FOXO) transcription factor family are profoundly involved in cell biology, including proliferation, hypertrophy, differentiation, DNA repair, cell cycle, energy recycling, and glucose metabolism.143
FOXO activation enhances the level of autophagy by directly binding to the promoter regions of autophagy-related genes. Specific FOXO deletion resulted in increased oxidative stress and apoptosis in osteoblasts, while FOXO3 overexpression prevented aging-related bone loss.144 Considering the role of autophagy in maintaining cell survival and the onset of aging-related physiological changes,145 it is natural to speculate that FOXO participates in the regulation of bone cell homeostasis via its induction effect on autophagy.
Activating transcription factor 4 (ATF4) from the cAMP-responsive element binding (CREB) protein family is also linked with both osteoblast function and autophagic activity. ATF4 is required in both the bone formation and terminal differentiation of osteoblasts. Mutation or disturbance in the level of ATF4 has been associated with two human genetic skeletal system diseases, Coffin–Lowry syndrome and neurofibromatosis type I.
ATF4 overexpression in fibroblasts could induce osteoblast-specific gene expression, osteocalcin synthesis, and aberrant mineral deposition.146 At the same time, ATF4 protects cells from amino acid starvation by enhancing the intakes of amino acids into cells. Interestingly, ATF4 is deemed an enhancer of cell survival and viability by upregulating the transcription of several autophagy genes, including microtubule-associated proteins 1A/1B light chain 3B (Map1lc3b) and Atg5. In addition, an aberrant increase in bone mass caused by neurofibromin-1 could be rescued by restricted amino acid intake.147
Autophagy takes part in the maintenance of osteocyte homeostasis
Autophagy is also essential when osteoblasts are incorporated into the bone matrix, thus terminally differentiating into osteocytes.148 Osteocytes are terminally differentiated cells embedded in the niches delimited by mineralized bone matrix. In contrast to the short cell lifespan of osteoblasts, osteocytes are very long-lived cells and are closer to neurons than other bone or cartilage cells in morphology.
First, osteoblasts undergo a drastic transition in cellular morphology and composition to become osteocytes, which requires active recycling of organelles.149 Second, with limited blood perfusion in the mineralized matrix, osteocytes are more susceptible to hypoxia and high oxidative stress150 and thus require a tighter budget on nutrient preservation. As discussed above, both of these aspects require active autophagy. Thus, not surprisingly, MAP1LC3 distribution is evident in a considerable proportion of embedded osteocytes, proving a basal level of autophagic activities.131
Specifically, osteocytes depend on autophagy to survive multiple adverse factors, including high ROS and hypoxia. When Atg7 was specifically knocked out in osteocytes using Dmp1 (dentin matrix protein 1)-Cre transgenic mice, oxidative stress increased as measured by ROS production and p66 phosphorylation. Moreover, this deletion could significantly lower the level of bone mass and bone remodeling, resulting in a phenotype that mimics the process of bone aging.151
Moreover, the present evidence suggests that osteocytes demonstrate greater autophagic activity than their progenitors. The expression level of LC3 in osteocytes is significantly higher than in osteoblasts.131 In addition, autophagy was correlated with the survival of osteocytes in a hypoxic environment. Pharmacological activation of autophagic activity reduced osteocyte apoptosis under high oxidative stress.148 In vivo-specific deletion of the Atg7 gene in osteocytes caused a significant reduction in bone mass, which was simultaneously associated with reduced osteoclast and osteoblast numbers and a disturbance of the balance between bone resorption and bone formation.151 Thus, autophagy plays a vital role in the survival of osteocytes and the maintenance of bone mass and remodeling.
In addition, in an ex vivo environment, autophagy in pre-osteocyte-like cells could be upregulated by nutrient starvation, hypoxia, or calcium stress,131 indicating the involvement of autophagy in the survival of osteocytes under stressed conditions. In addition, glucocorticoid treatment could increase the level of autophagic activity by up to 30-fold and profoundly influence osteocyte function.152
The major physiological function of osteocytes is to act as the mechanosensing system of skeletal tissue. The dendrite-like processes extending from osteocytes form a network and convert mechanical stimulus on the bone into biological signals that subsequently regulate the remodeling process. Primary cilium is an organelle that plays crucial roles in a variety of cellular functions, including mechanosensation.
Both primary cilium and autophagic activity have been tightly linked to various types of bone diseases, and their interaction has been suggested recently. Generally autophagic activity is suppressed in cells with shorter cilia. Conversely, when autophagy is downregulated either by exogenous pharmaceuticals or the intrinsic absence of autophagy-related proteins, ciliogenesis is enhanced and larger cilia length is observed. Further studies suggested that cilia and autophagic activity reciprocally regulate each other through the mTOR signaling pathway and the ubiquitin–proteasome system, and there might exist a negative feedback mechanism between autophagy and ciliogenesis.153
Aside from the mechanism directly mediated by primary cilium, mechanical stimulus may activate autophagy levels in MSCs, osteoblasts, and even chondrocytes. Mechanical loading-induced autophagy has been associated with survival and metabolism in various cell types.154 Cyclic changes in mechanical stimulus and its subsequent change in autophagy are indispensable to the network development of osteocytes.155
Autophagy is involved in bone resorption
Bone resorption is conducted by multinucleated osteoclasts, which are derived from hematopoietic stem cells upon stimulation with macrophage colony-stimulating factor (M-CSF) and RANKL (receptor activator of nuclear factor kappa-B ligand; also known as TNFSF11, tumor necrosis factor ligand superfamily member 11).114,156,157 When functioning, osteoclasts polarize to form a ruffled border at the cell-bone interface. Numerous sealed-off compartments are formed under the ruffled border, across which degradative enzymes are secreted onto the bony surface. The degraded bony matrix is then transported into osteoclasts via endocytosis for recycling.158 As mentioned above, autophagy is actively involved in both the differentiation and functioning of osteoclasts.132
Autophagy in the regulation of osteoclastogenesis
Osteoclasts originate from hematopoietic mononuclear myeloid stem cells, which in most cases reside in the marrow cavity. When the remodeling of the bony tissue is orchestrated and bone resorption is physiologically required or pathologically enforced, these mononuclear cells commit to a multinuclear osteoclastic phenotype and migrate onto the bony surfaces that are about to be remodeled.
The differentiation from adherent mononuclear cells into active osteoclasts is initiated by signal stimuli, including colony-stimulating factor 1 (CSF1) and TNFSF11/RANKL.159 At the initiation of osteoclastogenesis, mononuclear cells fuse with each other to become multinucleated giant cells.160 The recruitment of differentiating cells to the bone-remodeling site is dependent on chemokines, including CXCL12 (chemokine (C–X–C motif) ligand 12) and S1P (sphingosine 1-phosphate).
As mentioned above, autophagic activity upregulation protects cells from apoptosis. Under in vitro hypoxia and high oxidative stress conditions induced by glucocorticoids, the level of autophagic activity increased to reduce cell stress and thus protect osteoclast formation and survival.161–163 Similarly, under an in vivo environment, a regional hypoxic environment is expected, while differentiating osteoclasts migrate to the bone surface. Park et al.164 showed that hypoxia promoted increased expression of BNIP3, which consequently upregulated autophagic flux and MAP1LC3 recruitment to autophagosomes.165 Conversely, increased autophagic activity was correlated with enhanced osteoclast differentiation.163 These data suggested a role for the HIF1A-BNIP3 signaling pathway in mediating osteoclast differentiation.162
Autophagy and osteoclast functioning
When the bone-resorption program is activated, the terminally differentiated osteoclasts tightly adhere to the bony surface, and such attachment is achieved through specialized structures formed on the contact side of the osteoclasts named podosomes. Functional proteins, including actin filaments, F-actin, and actin monomers, serve as critical anchors for osteoclast attachment. The actual resorption is accomplished by the generation and secretion of lysosomes containing acids and proteases.
The lysosomes migrate to the ruffled border between osteoclasts and the bone surface, fuse with the cell membrane at the podosomes, and externalize the hydrochloric acid and proteases. The acids dissolve the mineral contents of the bone, and proteases, including matrix metalloproteinases, decompose the collagen matrix.166
When functioning, the active osteoclast needs to orchestrate the synthesis and externalization of acids and enzymes and the internalization and decomposition of the resolved matrix. In addition to their role in osteoclastogenesis, autophagy has been proven indispensable in the functioning of osteoclasts. When the exogenous autophagy inhibitor bafilomycin is added to the culture medium of osteoclasts, the resorptive activity is sharply decreased.167
The autophagy-related proteins ATG5, ATG7, ATG4B, and MAP1LC3 have all been suggested to play critical roles in the activation of resorption function. Both in vivo and in vitro data suggest that ATG5 and ATG7 promote osteoclastic functioning and guide lysosomes to target the actin ring.
Specific knockdown of genes related to autophagosome formation (Atg5, Atg7, Atg4b, or Lc3) in mononuclear osteoclast progenitors in mice leads to defects in lysosomal trafficking and formation of resorptive brush borders in osteoclasts and, consequently, downregulates bone-resorption activity and increases bone volume.132 Notably, neither ATG5 nor ATG7 affects osteoclastogenesis. In the specific knockdown models, the number of nuclei within osteoclasts, the presence of secretory lysosomes or even the expression of actin ring proteins was unaffected.
In addition, ATG4B modulation of MAPILC3A blocked both the resorptive activity and expression of CTSK (cathepsin K). When ATG5 was knocked down, significant downregulation in CDC42 activity and actin-ring disruption were observed. When MAPILC3A was knocked down, actin ring formation, resorption activity, and CTSK release were all inhibited.168 Consequently, it has been proposed that autophagy-related proteins can regulate bone-resorbing activity via the MAP1LC3-CDC42-dependent axis, which influences actin ring formation and ruffled border organization of osteoclasts.
In addition to the RANK/RANKL axis, inflammatory pathways are involved in the differentiation and function of osteoclasts. The presence of TNF-α activates NF-κb signaling and promotes osteoclastogenesis and bone resorption activities, and this mechanism is partially mediated through autophagy.169
As mentioned above, p62 is an important protein responsible for target sequestration for autophagosomes. In humans, the p62 mutation is observed in ~40%–50% of patients with Paget’s bone disease (PD), a condition involving increased bone resorption by osteoclasts, followed by excessive and disorganized bone formation by osteoblasts.170 This phenotype has also been recapitulated in mice, where a mutation in the gene Sqstm1 encoding the p62 protein results in excessive osteoclastic activity and a phenotype similar to Paget’s disease.171
Taken together, these results indicate that fine-tuning the balance of autophagic activity in all three types of bone cells is pivotal in the maintenance of bone homeostasis (Figs (Figs5,5, ,66).172

Autophagy and the functioning of three types of bone cells. Autophagy maintains the homeostasis of osteoblasts, osteoclasts, and osteocytes

Signal pathways regulating the bone-related autophagic activity. mTOR and ULK1 serve as pivots in conducting stress signals and growth factors to downstream autophagy-related proteins
reference link : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6804951/
More information: Marita Westhrin et al. Monoclonal immunoglobulins promote bone loss in multiple myeloma, Blood (2020). DOI: 10.1182/blood.2020006045



{kind=link}