









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



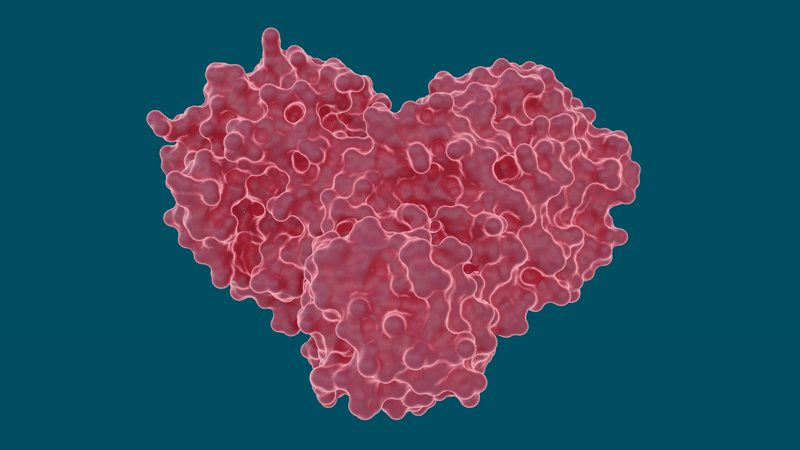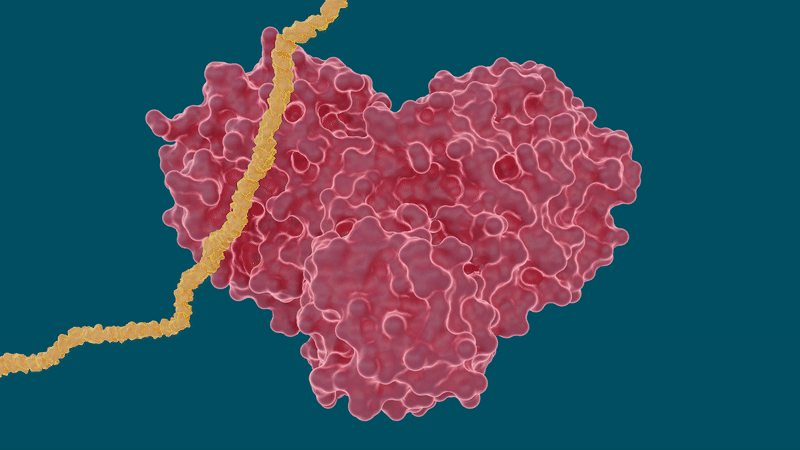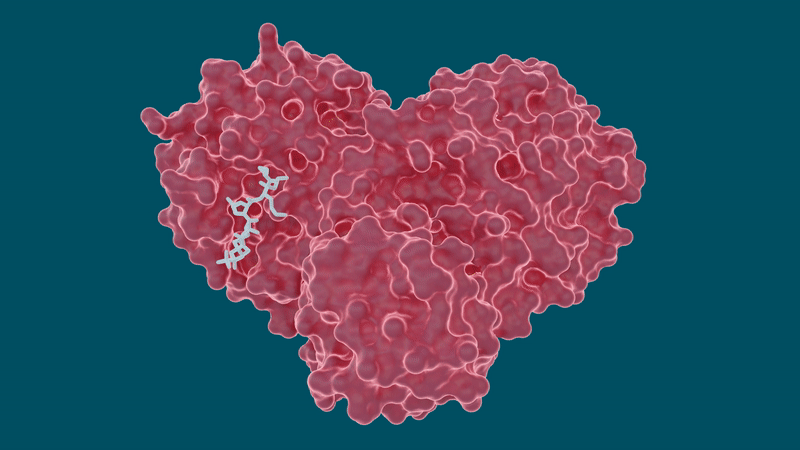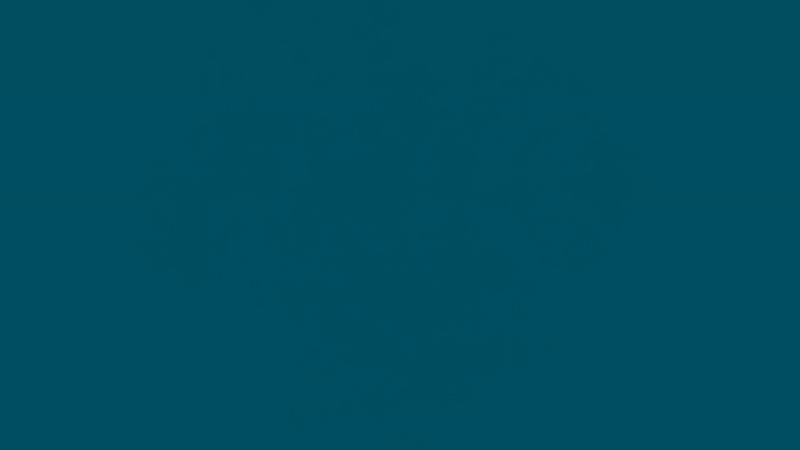
Esperimenti condotti da ricercatori presso l’Oak Ridge National Laboratory del Dipartimento dell’Energia hanno determinato che diversi farmaci per l’epatite C possono inibire la proteasi principale SARS-CoV-2, un enzima proteico cruciale che consente al nuovo coronavirus di riprodursi.
Inibire o bloccare il funzionamento di questa proteasi è fondamentale per impedire la diffusione del virus nei pazienti con COVID-19. Lo studio, pubblicato sulla rivista Structure, fa parte degli sforzi per sviluppare rapidamente trattamenti farmaceutici per COVID-19 riutilizzando farmaci esistenti noti per trattare efficacemente altre malattie virali.
“Attualmente, non ci sono inibitori approvati dalla Food and Drug Administration che prendono di mira la proteasi principale SARS-CoV-2 “, ha detto l’autore principale di ORNL Daniel Kneller.
“Quello che abbiamo scoperto è che i farmaci per l’epatite C si legano e inibiscono la proteasi del coronavirus. Questo è un primo passo importante per determinare se questi farmaci debbano essere considerati potenziali candidati riproposti per il trattamento di COVID-19 .
Il coronavirus SARS-CoV-2 si diffonde esprimendo lunghe catene di poliproteine che devono essere tagliate dalla proteasi principale per diventare proteine funzionali, rendendo la proteasi un importante bersaglio farmacologico per ricercatori e sviluppatori di farmaci.
Nello studio, il team ha esaminato diverse molecole farmacologiche ben note per potenziali sforzi di riproposizione, tra cui la leupeptina, un inibitore della proteasi naturale, e tre inibitori della proteasi dell’epatite C approvati dalla FDA: telaprevir, narlaprevir e boceprevir.
Il team ha eseguito misurazioni a raggi X a temperatura ambiente per costruire una mappa tridimensionale che ha rivelato come erano disposti gli atomi e dove si sono formati i legami chimici tra la proteasi e le molecole dell’inibitore del farmaco.
Gli esperimenti hanno prodotto risultati promettenti per alcuni farmaci per l’epatite C nella loro capacità di legare e inibire la proteasi principale SARS-CoV-2 , in particolare boceprevir e narlaprevir.
La leupeptina ha mostrato una bassa affinità di legame ed è stata esclusa come possibile candidato.
Per capire meglio quanto bene o quanto strettamente gli inibitori si legano alla proteasi, hanno utilizzato la cinetica enzimatica in vitro, una tecnica che consente ai ricercatori di studiare la proteasi e l’inibitore in una provetta per misurare l’affinità di legame dell’inibitore, o compatibilità, con il proteasi.
Maggiore è l’affinità di legame, più efficace è l’inibitore nel bloccare il funzionamento della proteasi.
“Quello che stiamo facendo è gettare le basi molecolari per questi potenziali inibitori che ripropongono farmaci rivelando la loro modalità di azione”, ha detto l’autore corrispondente di ORNL Andrey Kovalevsky.
“Mostriamo a livello molecolare come si legano, dove si legano e cosa stanno facendo alla forma dell’enzima. E, con la cinetica in vitro, sappiamo anche quanto bene si legano. Ogni pezzo di informazione ci avvicina di un passo alla comprensione di come fermare il virus “.
Lo studio fa anche luce su un comportamento peculiare della capacità della proteasi di cambiare o adattare la sua forma in base alle dimensioni e alla struttura della molecola inibitrice a cui si lega. Le tasche all’interno della proteasi in cui si attaccherebbe una molecola di farmaco sono altamente malleabili o flessibili e possono aprirsi o chiudersi in una misura a seconda delle dimensioni delle molecole del farmaco.
Prima della pubblicazione del documento, i ricercatori hanno reso i loro dati pubblicamente disponibili per informare e assistere le comunità scientifiche e mediche. Sono necessarie ulteriori ricerche, inclusi studi clinici, per convalidare l’ efficacia e la sicurezza dei farmaci come trattamento COVID-19.
“La ricerca suggerisce che vale la pena considerare gli inibitori dell’epatite C come potenziali candidati riproposti.
Il rilascio immediato dei nostri dati consente alla comunità scientifica di iniziare a esaminare le interazioni tra questi inibitori e la proteasi “, ha affermato l’autore corrispondente dell’ORNL Leighton Coates.
“Non è possibile progettare un farmaco senza sapere come funziona a livello molecolare e i dati che forniamo sono esattamente ciò di cui gli sviluppatori hanno bisogno per progettare farmaci più forti e più strettamente vincolanti per trattamenti più efficaci.”
I farmaci anti-coronavirus disponibili per via orale sarebbero altamente desiderabili, in quanto possono essere somministrati in ambiente ambulatoriale, cioè senza richiedere il ricovero in ospedale. In quanto tali, possono essere somministrati al momento della diagnosi, il che potrebbe impedire la progressione verso una malattia più grave o protratta e quindi ridurre notevolmente la morbilità e la mortalità.
Tuttavia, l’unico farmaco antivirale di provata efficacia contro SARSCoV2, il remdesivir, è un farmaco per via endovenosa che può essere somministrato solo a pazienti ospedalizzati.
Anche allora riduce la durata della malattia e il rischio di morte solo di circa il 30% ciascuno (3).
Il desametasone si è dimostrato efficace nel migliorare la sopravvivenza nel COVID-19 in fase avanzata sopprimendo le risposte iperinfiammatorie tardive che causano insufficienza respiratoria e d’organo piuttosto che inibendo la replicazione virale (4). Tuttavia, è inefficace se somministrato prima (4) e può anche essere dannoso perché inibisce le risposte immunitarie dirette contro il virus (5-7).
Pertanto, non esistono farmaci in grado di sopprimere efficacemente la replicazione del coronavirus in ambiente ambulatoriale durante l’intero periodo post-infezione, ma sono urgentemente necessari.
Gli inibitori della proteasi principale del coronavirus (Mpro), una proteina chiave nel ciclo di vita del coronavirus, potrebbero essere particolarmente efficaci come agenti antivirali (8, 9).
La Mpro (chiamata anche proteasi simile a 3C per la sua omologia con la proteasi 3C dell’enterovirus) è una proteasi cisteina che scinde la grande poliproteina non strutturale del coronavirus in pezzi più piccoli per assemblare la RNA replicasi (10).
Gli inibitori sperimentali del coronavirus Mpro proteggono topi e gatti dalla malattia letale del coronavirus, convalidando Mpro come bersaglio medicinale (11-13).
Vi è attualmente un intenso interesse per la scoperta di inibitori efficaci per SARSCoV2 Mpro. Poiché le specie di coronavirus Mpro condividono tutte profili di specificità del substrato simili (14), gli inibitori precedentemente caratterizzati per altri coronavirus hanno, non sorprende, dimostrato attività inibitoria anche su SARSCoV2 Mpro. Ad esempio, il composto accettore di Michael N3, sviluppato nel 2005 come inibitore di SARSCoV1 Mpro (15), inibisce anche il 96% di SARSCoV2 Mpro identico in vitro (15, 16, 16).
Una serie di inibitori a base di chetoamide sviluppati per SARSCoV1 Mpro sono risultati efficaci anche contro SARSCoV2 Mpro in vitro (17) e hanno dato origine all’inibitore SARSCoV2 Mpro modificato 13b (18). Infine, GC376, un profarmaco aldeidico ad ampio spettro di varie specie di coronavirus Mpro e 11a, un composto aldeidico progettato per inibire SARSCoV1 Mpro, sono stati trovati per inibire SARSCoV2 Mpro (19, 20).
Tuttavia, non è chiaro se gli inibitori noti del coronavirus Mpro abbiano gli attributi necessari per essere terapie orali sicure ed efficaci per le malattie del coronavirus umano. In 11a e nella forma attiva di GC-376, un gruppo reattivo aldeidico (testata) reagisce con il sito attivo cisteina, ma le aldeidi sono generalmente considerate farmaci candidati non ideali a causa della tossicità causata da reazioni fuori bersaglio (21-24).
Ad esempio, è stato dimostrato che l’elevata reattività delle testate aldeidiche guida l’inibizione aspecifica delle proteasi della cisteina (25). Gli inibitori della proteasi con chetoamide e testate di accettore di Michael possono mostrare una specificità, stabilità e biodisponibilità orale migliori rispetto a quelli con aldeidi (26-29), e gli esempi sono progrediti fino a studi clinici in fase avanzata o approvazione normativa (30, 31).
I composti 13b e N3 utilizzano rispettivamente testate chetoamide e accettore di Michael, ma sembrano avere un’efficacia insufficiente per essere terapeuticamente utili.
Mentre 13b dimostra una concentrazione inibitoria del 50% (IC50) su Mpro in vitro di 670 nM, la concentrazione effettiva del 50% (EC50) nel bloccare la replicazione di SARSCoV2 nelle cellule umane è ∼4 μM (18).
Ciò si confronta sfavorevolmente con la concentrazione raggiunta nei polmoni di topo di <0,1 μM (18). Per N3, mentre mostra una bassa IC50 in vitro, la sua EC50 per la replicazione di SARSCoV2 è di 17 μM (16), indicando una scarsa permeabilità cellulare. Ciò non sorprende date le sue grandi dimensioni (mw 681) e numerosi gruppi polari. Pertanto, lo sviluppo di nuovi modelli di inibitori di Mpro con bassa tossicità e bassa EC50 nelle cellule rimane una priorità urgente.
Più inibitori della proteasi clinicamente approvati incorporano strutture cicliche per migliorare l’affinità pre-organizzando gli inibitori in una conformazione favorevole al legame (32).
In termini energetici, questo è considerato pagare la penalità entropica della rigidità conformazionale prima del legame (33, 34), ed è stato sperimentalmente confermato che la ciclizzazione aumenta l’affinità dell’inibitore della proteasi (35).
In questo studio, esploriamo la capacità di una porzione ciclica di migliorare l’affinità degli inibitori del coronavirus Mpro. Abbiamo sviluppato un nuovo inibitore, ML1000, con peso molecolare di 549 Da e valore IC50 di 12 nM su SARSCoV2 Mpro.
ML1000 inibisce la replicazione di SARSCoV2 nelle cellule umane con un EC50 di 0,1 µM, e quindi è l’inibitore Mpro non aldeidico più potente riportato finora. ML1000 rappresenta una nuova classe strutturale di potenti inibitori della chetoamide del coronavirus Mpro e può servire come punto di partenza promettente per lo sviluppo di farmaci candidati contro le malattie del coronavirus umano.
RISULTATI
Durante la visualizzazione della struttura co-cristallina di 13b e SARSCoV2 Mpro (18), abbiamo notato che 13b adotta una piega pronunciata nella sua catena principale al residuo P2, cioè il 2 ° residuo N-terminale al legame scissile rivolto verso la tasca S2 dell’enzima (Fig. 1A).
A causa del nostro precedente lavoro con la proteasi dell’HCV e i suoi inibitori delle piccole molecole (36), abbiamo riconosciuto che questo nodo è simile a quello creato dagli anelli dell’analogo della prolina nei farmaci anti-HCV approvati clinicamente boceprevir, narlaprevir e telaprevir (Fig. 1B -D).
Come 13b, questi farmaci sono inibitori covalenti a base di chetoamide, che servono come substrati per l’attacco nucleofilo da parte della cisteina del sito attivo deprotonato.

inibitori della proteasi dell’HCV con un analogo della prolina P2 possono essere agganciati al sito attivo SARSCoV2 Mpro. (A) Struttura co-cristallina di SARSCoV2 Mpro e inibitore 13b (PDB 6Y2G). (B) Utilizzando Pymol, boceprevir è stato inserito nel sito attivo SARSCoV2 Mpro e i legami non vincolati sono stati ruotati manualmente per un complementare ottimale con le tasche S1 e S2 e il legame idrogeno con il carbonile della spina dorsale di Glu-166. (C) Telaprevir è stato ancorato in modo simile al sito attivo SARSCoV2 Mpro per un complementare ottimale con le tasche S1, S2 e S4 e il legame idrogeno con il carbonile della spina dorsale di Glu-166. (D) L’allineamento del cocristallo 13b-Mpro con la struttura di boceprevir ancorata manualmente mostra che la spina dorsale del segmento analogo P2 di 13b è sovrapponibile con l’analogo prolina di boceprevir.
L’attracco rigido manuale di boceprevir ha mostrato che potrebbe adattarsi al sito attivo di SARSCoV2 Mpro con una buona complementarità di forma dai suoi gruppi P1, P2 e P4 (Fig. 1B).
Inoltre, il gruppo urea di boceprevir alla giunzione P3-P4 potrebbe impegnarsi in un legame idrogeno bidentato con la spina dorsale carbonile di Mpro Glu-166.
Anche il derivato di boceprevir narlaprevir si è dimostrato complementare nei suoi gruppi P1 e P2, che sono identici a boceprevir, ma il suo gruppo P4 era chiaramente troppo grande per la tasca S4. Telaprevir potrebbe anche essere ancorato manualmente con una buona complementarità del suo sidechain P1 e del suo gruppo P2, che è un analogo della prolina diverso da quello di boceprevir (Fig. 1C), mentre il suo gruppo P4 appariva leggermente troppo grande per la tasca S4.
Il gruppo P3 dei substrati del coronavirus Mpro è rivolto verso la soluzione (37), così come il gruppo t-butile nella posizione analoga di boceprevir, telaprevir e narlaprevir. È interessante notare che gli angoli φ e Ψ dell’anello prolina in boceprevir hanno ripercorso con precisione gli atomi della spina dorsale di 13b nella configurazione legata (Fig. 1D).
Abbiamo quindi ipotizzato che boceprevir e, in misura minore, narlaprevir e telaprevir, possano essere in grado di inibire SARSCoV2 Mpro.
Per affrontare questa ipotesi, abbiamo eseguito una rapida valutazione preliminare utilizzando un saggio di cellule vive della funzione Mpro. Abbiamo co-espresso SARSCoV2 Mpro e una proteina substrato comprendente la proteina fluorescente verde (GFP), un sito substrato e la sequenza CAAX che mira alla membrana (Fig. 2A).
La scissione nella sequenza del substrato libera GFP, consentendo la quantificazione dell’attività mediante immunoblotting. Potremmo quindi valutare la capacità dei farmaci di inibire l’attività di Mpro nelle cellule. Nelle cellule HEK293A, abbiamo scoperto che boceprevir e telaprevir inibiscono SARSCoV2 Mpro a concentrazioni micromolari (Fig. 2B).
Questi risultati hanno fornito la prova della capacità degli inibitori dell’HCV con un gruppo prolina P2 di inibire SARSCoV2 Mpro.

Evidenza preliminare dell’attività dell’inibitore della proteasi dell’HCV su SARSCoV2 Mpro. (A) Schema del costrutto reporter della proteasi. Per questo esperimento è stata utilizzata una versione immatura di Mpro con attività ridotta per imitare più stati naturali di Mpro. Durante l’esperimento, è previsto che Mpro si attivi automaticamente rimuovendo le estensioni del terminale. (B) L’immunoblot di lisati da cellule HEK293A transitoriamente transfettate con il costrutto reporter mostrato in (A) e cresciute in assenza o presenza di diverse concentrazioni di telaprevir o boceprevir mostra la soppressione della scissione del substrato a concentrazioni di farmaco micromolari.
Successivamente abbiamo eseguito un test pilota sulla capacità degli inibitori dell’HCV con un gruppo prolina P2 di inibire SARSCoV2 Mpro in vitro (Fig. 3A).
Come controlli, abbiamo anche testato GC-376 come un noto inibitore del coronavirus Mpro ad ampio spettro, ebselen e disulfiram come composti recentemente segnalati per inibire SARSCoV2 Mpro (16) e ritonavir come inibitore della proteasi senza alcuna omologia strutturale ai substrati del coronavirus Mpro. In effetti, abbiamo osservato che boceprevir, narlaprevir, telaprevir, GC-376, disulfiram ed ebselen (a una concentrazione di 150 μM in tampone equilibrato con aria) erano in grado di inibire SARSCoV2 Mpro, mentre ritonavir non mostrava alcun effetto inibitorio (Fig. 3A).
Per inciso, il disulfiram e l’ebselen non hanno mostrato alcun effetto inibitorio in presenza di DTT, mentre altri composti non sono stati influenzati dal DTT. Questi risultati sono coerenti con le proposte di disulfiram ed ebselen che formano un legame riducibile con Mpro (38).
Poiché l’interno della cellula si sta riducendo, la riduzione degli addotti di ebselen potrebbe contribuire alla sua EC50 relativamente alta di 4,7 μM per bloccare la replicazione di SARSCoV2 nelle cellule (16).

Abbiamo quindi eseguito curve di inibizione completa SARSCoV2 Mpro per ottenere i valori IC50 per i composti di cui sopra. Il test pilota iniziale sopra è stato eseguito con SARSCoV2 Mpro con un tag His6 C-terminale per una rapida purificazione, ma la presenza di un’estensione C-terminale può avere un effetto inibitorio sull’attività della proteasi (39, 40). Abbiamo quindi eseguito curve di inibizione su SARSCoV2 Mpro sia C-terminale esteso che completamente maturo (Fig. 3B).
Abbiamo testato farmaci in presenza di DTT, ad eccezione di ebselen e disulfiram, per i quali abbiamo omesso il DTT. I nostri risultati hanno mostrato che gli inibitori della proteasi dell’HCV contenenti prolina boceprevir, telaprevir e narlaprevir potrebbero inibire l’attività in una certa misura sia nelle forme estese C-terminale che in quelle completamente mature di SARSCoV2 Mpro. Tra questi, boceprevir era il più potente, con un valore IC50 di 4,1 μM su SARSCoV2 Mpro maturo (Fig. 3B, Tabella 1).

Avendo stabilito che gli analoghi della prolina P2 in boceprevir, telaprevir e narlaprevir erano compatibili con il legame al sito attivo SARSCoV2 Mpro, abbiamo successivamente cercato di progettare inibitori del coronavirus di prossima generazione che potessero combinare la stabilizzazione entropica conferita da questi anelli P2 con gruppi laterali ottimizzati per il legame al coronavirus Mpro. Un residuo P1 Gln è fortemente preferito da tutte le specie di coronavirus Mpro (14).
In effetti, questa preferenza è conservata con le proteasi correlate dell’enterovirus 3C come la proteasi del rhinovirus umano (HRV) e anche con le proteasi del potyvirus più lontane come la proteasi del virus dell’etch del tabacco (TEV) (41, 42). A partire dagli inibitori della proteasi HRV AG7088 (rupintrivir) e dal suo derivato AG7404 (43, 44), gli inibitori della proteasi dell’enterovirus e del coronavirus hanno incorporato un gruppo γ-lattamico per imitare la funzione dell’accettore del legame idrogeno di Gln nella posizione P1.
Le strutture cocristalline degli inibitori enzimatici hanno confermato che il gruppo γ-lattammico è posizionato in modo simile al gruppo Gln ammide nelle strutture naturali (37, 45). Abbiamo quindi progettato un nuovo inibitore, ML1000, con la testata chetoamide di boceprevir, il gruppo γ-lattamil P1 di rupintrivir e le frazioni da P2 a P4 (inclusi spina dorsale e catene laterali) di boceprevir (Fig. 4A).
Abbiamo anche progettato un secondo inibitore, ML1100, composto dalla testata chetoamide di boceprevir, il gruppo γ-lattamil P1 di rupintrivir, la struttura ciclica P2 di telaprevir e le frazioni P3 e P4 di boceprevir (Fig. 4A).
Abbiamo mantenuto la struttura P3 e P4 di boceprevir poiché questi gruppi non hanno mostrato alcuna collisione con la sacca enzimatica nel modello di boceprevir ancorato manualmente e poiché la loro idrofobicità probabilmente contribuisce alla permeabilità della membrana.

Progettazione e potenza in vitro di un nuovo inibitore del coronavirus Mpro. (A) Strutture di boceprevir, telaprevir, ML1000 e ML1100. ML1000 è essenzialmente boceprevir con un gruppo γ-lattamile al posto del gruppo ciclobutanile P1. ML1100 sostituisce l’analogo biciclico della prolina P2 di boceprevir con quello di telaprevir. (B) Misurazioni IC50 di ML1000 e ML1100 con SARSCoV2 Mpro maturo a 100 nM. Con una concentrazione enzimatica a 100 nM, l’IC50 più basso possibile che può essere rilevato in teoria è 50 nM. Pertanto, i valori di IC50 sono stati misurati anche per ML1000 e GC-376 con enzima 20 nM. (C) I valori IC50 di ML1000 e GC-376 misurati con SARSCoV2 Mpro maturo da 20 nM erano rispettivamente di 12 e 14 nM. Pertanto, ML1000 e GC-376 mostrano uno stretto legame, anche a 20 nM di Mpro, ei valori di IC50 possono essere ancora limitati dalla concentrazione dell’enzima. Vengono mostrati i valori medi di 3 esperimenti indipendenti. Le barre di errore rappresentano la deviazione standard.
ML1000 e ML1100 si sono dimostrati inibitori molto potenti di SARSCoV2 Mpro. In presenza di 100 nM di enzima, ML1000 e ML1100 hanno prodotto valori IC50 di 34 nM e 147 nM (Fig. 4B, Tabella 1), rispetto a 54 nM per GC-376 (Fig. 3B, Tabella 1).
Contro l’enzima Mpro del virus dell’epatite murina coronavirus (MHV) lontanamente imparentato, ML1000 e ML1100 hanno mostrato valori IC50 di 130 e 301 nM, rispettivamente, rispetto a 67 nM per GC-376 (Tabella 1, Figura di supporto 1).
Questi risultati dimostrano che gli inibitori della chetoamide con analoghi dell’anello prolina in posizione P2 possono funzionare come inibitori del coronavirus Mpro ad ampio spettro con una potenza che si avvicina a quella degli inibitori dell’aldeide.

Abbiamo notato che i valori IC50 misurati di ML1000 e GC-376 per SARSCoV2 Mpro erano entro l’errore sperimentale del limite teorico della metà della concentrazione enzimatica nel test (46), ostacolando la capacità di discernere le differenze di potenza tra di loro. Abbiamo quindi misurato anche i valori IC50 a una concentrazione inferiore di SARSCoV2 Mpro di 20 nM (Fig. 4C).
In queste condizioni, ML1000 e GC-376 mostravano ancora valori di IC50 comparabili di 12 e 14 nM, rispettivamente, al di sotto della concentrazione di Mpro (cioè il limite di legame stretto).
La temperatura fisiologica, rispetto ai 30 ° C a cui vengono solitamente eseguiti questi test, potrebbe potenzialmente aumentare la flessibilità strutturale e alterare l’affinità farmacologica di Mpro. Tuttavia, non abbiamo trovato differenze significative di potenza tra 37 ° C e 30 ° C per ciascun inibitore (Tabella 1, Figura di supporto 2).
Questi risultati indicano che ML1000 ha un’elevata potenza in vitro e rappresenta l’inibitore SARSCoV2 Mpro non aldeidico più stretto scoperto finora.

Successivamente, abbiamo testato l’efficacia dei nuovi inibitori contro SARSCoV2 Mpro in cellule Huh7 umane misurando l’entità della scissione del substrato coespresso (Fig. 5). Qui, abbiamo osservato che l’ordine dei ranghi degli effettivi è GC-376> ML1000> ML1100> boceprevir (Fig. 5).
Va sottolineato che questo test non misura direttamente la frazione di Mpro attivo. Se una piccola frazione di Mpro attivo è sufficiente per provocare la scissione del substrato, ciò aumenterebbe effettivamente la concentrazione misurata necessaria per sopprimere l’attività della proteasi.
Tuttavia, il test fornisce una valutazione della capacità relativa di un insieme di farmaci di attraversare la membrana cellulare e inibire l’Mpro all’interno delle cellule viventi.

Infine, abbiamo testato la capacità di ML1000 e ML1100 di inibire la replicazione del virus SARSCoV2 nelle cellule intestinali umane Caco-2. Per confronto, abbiamo testato boceprevir e GC-376. Boceprevir ha inibito la replicazione virale con un EC50 di 0,2 µM, mentre GC-376 era ancora più potente, con un EC50 notevolmente basso di 0,1 nM (Tabella 2, Fig. 6). ML1000 e ML1100 hanno inibito la replicazione di SARSCoV2 con valori EC50 rispettivamente di 0,1 e 0,2 µM (Tabella 2, Fig. 6).
Sebbene il miglioramento dell’efficacia antivirale di ML1000 rispetto a boceprevir non sia grande quanto il miglioramento dell’inibizione enzimatica osservato in vitro (Tabella 1), il valore EC50 di ML1000 è nondimeno il più basso riportato fino ad oggi per qualsiasi inibitore non aldeidico di SARSCoV2 Mpro.
Inoltre, ML1000 e ML1100 non hanno mostrato citotossicità alla dose più alta testata di 100 μM nelle cellule Caco-2 e Huh-7 (Tabella 2, Figura di supporto 3), con conseguenti indici di selettività rispettivamente di> 1000 e> 500. Questi risultati confermano che ML1000 è un potente e selettivo inibitore della chetoamide della replicazione di SARSCoV2.



RIFERIMENTI
REFERENCES
- 1.M. Letko, S. N. Seifert, K. J. Olival, R. K. Plowright, V. J. Munster, Batborne virus diversity, spillover and emergence. Nat Rev Microbiol 18, 461–471 (2020). Google Scholar
- 2.W. H. Organization, Weekly Operational Update on COVID19, 28 August 2020. Google Scholar
- 3.J. H. Beigel et al., Remdesivir for the Treatment of Covid19 Preliminary Report. N Engl J Med (2020). Google Scholar
- 4.C. G. Recovery et al., Dexamethasone in Hospitalized Patients with Covid19 Preliminary Report. N Engl J Med (2020). Google Scholar
- 5.D. W. Cain, J. A. Cidlowski, After 62 years of regulating immunity, dexamethasone meets COVID19. Nat Rev Immunol (2020). Google Scholar
- 6.M. A. Lim, R. Pranata, Worrying situation regarding the use of dexamethasone for COVID19. Ther Adv Respir Dis 14, 1753466620942131 (2020). Google Scholar
- 7.T. C. Theoharides, P. Conti, Dexamethasone for COVID19? Not so fast. J Biol Regul Homeost Agents 34, (2020). Google Scholar
- 8.X. Deng et al., Coronaviruses resistant to a 3Clike protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 88, 11886–11898 (2014). Abstract/FREE Full TextGoogle Scholar
- 9.S. Ullrich, C. Nitsche, The SARSCoV2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377 (2020). Google Scholar
- 10.Y. Kim et al., Broadspectrum antivirals against 3C or 3Clike proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 86, 11754–11762 (2012). Abstract/FREE Full TextGoogle Scholar
- 11.Y. Kim et al., Broadspectrum inhibitors against 3Clike proteases of feline coronaviruses and feline caliciviruses. J. Virol. 89, 4942–4950 (2015). Abstract/FREE Full TextGoogle Scholar
- 12.Y. Kim et al., Reversal of the Progression of Fatal Coronavirus Infection in Cats by a BroadSpectrum Coronavirus Protease Inhibitor. PLoS Pathog 12, e1005531 (2016). CrossRefPubMedGoogle Scholar
- 13.N. C. Pedersen et al., Efficacy of a 3Clike protease inhibitor in treating various forms of acquired feline infectious peritonitis. J Feline Med Surg 20, 378–392 (2018). CrossRefPubMedGoogle Scholar
- 14.C. P. Chuck, H. F. Chow, D. C. Wan, K. B. Wong, Profiling of substrate specificities of 3Clike proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One 6, e27228 (2011). CrossRefPubMedGoogle Scholar
- 15.H. Yang et al., Design of widespectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3, e324 (2005). CrossRefPubMedGoogle Scholar
- 16.Z. Jin et al., Structure of Mpro from COVID19 virus and discovery of its inhibitors. Nature 582, 289–293 (2020). Google Scholar
- 17.L. Zhang et al., αKetoamides as BroadSpectrum Inhibitors of Coronavirus and Enterovirus Replication: StructureBased Design, Synthesis, and Activity Assessment. J. Med. Chem. 63, 4562–4578 (2020). Google Scholar
- 18.L. Zhang et al., Crystal structure of SARSCoV2 main protease provides a basis for design of improved αketoamide inhibitors. Science 368, 409–412 (2020). Abstract/FREE Full TextGoogle Scholar
- 19.W. Dai et al., Structurebased design of antiviral drug candidates targeting the SARSCoV2 main protease. Science 368, 1331–1335 (2020). Abstract/FREE Full TextGoogle Scholar
- 20.W. Vuong et al., Feline coronavirus drug inhibits the main protease of SARSCoV2 and blocks virus replication. Nat Commun 11, 4282 (2020). Google Scholar
- 21.K. Gluza, P. Kafarski, Transition state analogues of enzymatic reaction as potential drugs. Drug Discovery (2013). Google Scholar
- 22. S. L. Harer, M. S. Bhatia, N. M. Bhatia, Proteasome inhibitors mechanism; source for design of newer therapeutic agents. J Antibiot (Tokyo) 65, 279–288 (2012). PubMedGoogle Scholar
- 23.M. Siklos, M. BenAissa, G. R. J. Thatcher, Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharmaceutica Sinica B (2015). Google Scholar
- 24.Vasudevan et al., Covalent binders in drug discovery. Prog. Med. Chem. 58, 1–62 (2019). Google Scholar
- 25.L. Zhu et al., Peptide aldehyde inhibitors challenge the substrate specificity of the SARScoronavirus main protease. Antiviral Res 92, 204–212 (2011). CrossRefPubMedWeb of ScienceGoogle Scholar
- 26.D. G. Barrett et al., Orally bioavailable small molecule ketoamidebased inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 14, 2543–2546 (2004). PubMedGoogle Scholar
- 27.K. O. Chang, Y. Kim, W. C. Groutas, D. Hua, S. Lj, Broadspectrum antivirals against 3c or 3clike proteases of picornaviruslike supercluster: picornaviruses, caliciviruses and coronaviruses. US Patent 9,474,759 (2016). Google Scholar
- 28. Y. Kim et al., Potent inhibition of enterovirus D68 and human rhinoviruses by dipeptidyl aldehydes and αketoamides. Antiviral Res 125, 84–91 (2016). Google Scholar
- 29.Y. Shirasaki et al., Exploration of orally available calpain inhibitors 2: peptidyl hemiacetal derivatives. J. Med. Chem. 49, 3926–3932 (2006). PubMedGoogle Scholar
- 30.S. Amslinger, The tunable functionality of α, βunsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem (2010). Google Scholar
- 31.F. Sutanto, M. Konstantinidou, A. Dömling, Covalent inhibitors: a rational approach to drug discovery. RSC Medicinal Chemistry (2020). Google Scholar
- 32.D. I. Soumana et al., Structural and Thermodynamic Effects of Macrocyclization in HCV NS3/4A Inhibitor MK5172. ACS Chem Biol 11, 900–909 (2016). CrossRefGoogle Scholar
- 33.Z. Fang, Y. Song, P. Zhan, Q. Zhang …, Conformational restriction: an effective tactic in’followon’based drug discovery. Future Medicinal Chemistry (2014). Google Scholar
- 34.P. Y. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge …, Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. … (1994). Google Scholar
- 35.K. Ghosh, G. E. Schiltz, L. N. Rusere …, Design and synthesis of potent macrocyclic HIV1 protease inhibitors involving P1–P2 ligands. Organic … (2014). Google Scholar
- 36.C. L. Jacobs, R. K. Badiee, M. Z. Lin, StaPLs: versatile genetically encoded modules for engineering druginducible proteins. Nat Methods 15, 523–526 (2018). Google Scholar
- 37.M. F. Hsu et al., Mechanism of the maturation process of SARSCoV 3CL protease. J. Biol. Chem. 280, 31257–31266 (2005). Abstract/FREE Full TextGoogle Scholar
- 38.H. Sies, M. J. Parnham, Potential therapeutic use of ebselen for COVID19 and other respiratory viral infections. Free Radic Biol Med 156, 107–112 (2020). Google Scholar
- 39.T. Muramatsu et al., Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3Clike protease (SARSCoV 3CLpro) from its polyproteins. FEBS J 280, 2002–2013 (2013). CrossRefPubMedGoogle Scholar
- 40.X. Xue et al., Production of authentic SARSCoV M(pro) with enhanced activity: application as a novel tagcleavage endopeptidase for protein overproduction. J. Mol. Biol. 366, 965–975 (2007). CrossRefPubMedGoogle Scholar
- 41.M. J. Adams, J. F. Antoniw, F. Beaudoin, Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol Plant Pathol 6, 471–487 (2005). CrossRefPubMedWeb of ScienceGoogle Scholar
- 42.J. Tan et al., 3C protease of enterovirus 68: structurebased design of Michael acceptor inhibitors and their broadspectrum antiviral effects against picornaviruses. J. Virol. 87, 4339–4351 (2013). Abstract/FREE Full TextGoogle Scholar
- 43.D. A. Matthews et al., Structureassisted design of mechanismbased irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A. 96, 11000–11007 (1999). Abstract/FREE Full TextGoogle Scholar
- 44.K. Patick et al., In vitro antiviral activity and singledose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 49, 2267–2275 (2005). Abstract/FREE Full TextGoogle Scholar
- 45.S. Yang et al., Synthesis, crystal structure, structureactivity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 49, 4971–4980 (2006). CrossRefPubMedGoogle Scholar
- 46.R. Z. Cer, U. Mudunuri, R. Stephens, F. J. Lebeda, IC50toKi: a webbased tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 37, W441–5 (2009). CrossRefPubMedGoogle Scholar
- 47.B. R. Bacon et al., Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364, 1207–1217 (2011). CrossRefPubMedWeb of ScienceGoogle Scholar
- 48.L. Fu et al., Both Boceprevir and GC376 efficaciously inhibit SARSCoV2 by targeting its main protease. Nat Commun 11, 4417 (2020). Google Scholar
- 49.C. Ma et al., Boceprevir, GC376, and calpain inhibitors II, XII inhibit SARSCoV2 viral replication by targeting the viral main protease. Cell Res 30, 678–692 (2020). Google Scholar
- 50.B. J. Anson et al., Broadspectrum inhibition of coronavirus main and papainlike proteases by HCV drugs. researchsquare.com (2020). Google Scholar
- 51.X. Zuo et al., Expression and purification of SARS coronavirus proteins using SUMOfusions. Protein Expr Purif 42, 100–110 (2005). CrossRefPubMedGoogle Scholar
More information: Daniel W. Kneller et al, Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals, Structure (2020). DOI: 10.1016/j.str.2020.10.007



{kind=link}