










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Many cancer researchers can claim to have devised “smart bombs.”
What has been missing is the stealth bomber – a delivery system that can slip through the body’s radar defenses.
Oncolytic viruses, or viruses that preferentially kill cancer cells, have been discussed and tested for decades.
An oncolytic virus against melanoma was approved by the FDA in 2015. But against metastatic cancers, they’ve always faced an overwhelming barrier: the human immune system, which quickly captures viruses injected into the blood and sends them to the liver, the body’s garbage disposal.
Researchers at Emory and Case Western Reserve have now circumvented that barrier. They’ve re-engineered human adenovirus, so that the virus is not easily caught by parts of the innate immune system.
This makes it possible to inject the virus into the blood, without arousing a massive inflammatory reaction.
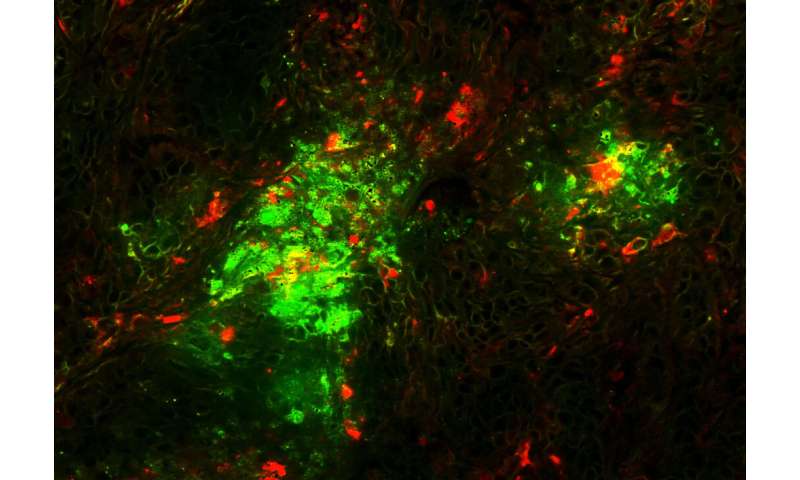
A cryo-electron microscopy structure of the re-engineered virus and the virus’s ability to eliminate disseminated tumors in mice are reported in Science Translational Medicine.
“The innate immune system is quite efficient at sending viruses to the liver when they are delivered intravenously,” says lead author Dmitry Shayakhmetov, Ph.D.
“For this reason, most oncolytic viruses are delivered directly into the tumor, without affecting metastases. In contrast, we think it will be possible to deliver our modified virus systemically at doses high enough to suppress tumor growth—without triggering life-threatening systemic toxicities.”
The co-first authors of the Science Translational Medicine paper are Emory associate scientist Svetlana Atasheva, Ph.D. and Case Western Reserve graduate student Corey Emerson. Shayakhmetov is professor of medicine and pediatrics at Emory University School of Medicine and a member of Lowance Center for Human Immunology and Emory Vaccine Center.
Shayakhmetov has been working for 15 years with structural biologist Phoebe Stewart, Ph.D., professor in the Department of Pharmacology and a member of Cleveland Center for Membrane and Structural Biology at Case Western Reserve University. Their focus: re-engineering adenovirus, a delivery system that has been used in dozens of cancer clinical trials to stimulate host anti-tumor response.
Adenoviruses have also been central to gene therapy studies. Shayakhmetov recalls the 1999 death of Jesse Gelsinger, a volunteer in a gene therapy clinical trial who died of cytokine storm and multi-organ failure connected with high doses of an adenovirus vector delivered into the bloodstream.
He says that event inspired him to retool adenovirus, so that it would not set off a strong inflammatory reaction.
He views the re-engineered adenovirus as a platform technology, which can be adapted and customized for many types of cancer, and even to individual cancer patients as a form of personalized cancer therapy.
“This is a new avenue for treatment of metastatic cancers,” Shayakhmetov says.
“You can arm it with genes and proteins that stimulate immunity to cancer, and you can assemble the capsid, a shell of the virus, like you’re putting in Lego blocks.”

Shayakhmetov started working on the modified virus technology while he was at the University of Washington and founded a company, AdCure Bio, to bring a potentially life-saving therapy to patients with metastatic disease.
In 2012, Shayakhmetov’s and Stewart’s labs published a cryo-EM analysis of how adenovirus interacts with one host factor in the blood, coagulation factor X, in Science.
“Sometimes even small changes in structural proteins can be catastrophic and prevent assembly of the infectious virus,” Stewart says.
“In this case, we modified adenovirus in three places to minimize virus interactions with specific blood factors. We found that the virus still assembles and remains functional for infecting and killing tumor cells.”
It is still possible for a slower-building adaptive immune response to develop to the modified virus, similar to that observed with a vaccine. A panel of viruses could be used for sequential administration to cancer patients to extend therapeutic benefits, Shayakhmetov says.
“Our study is the first to show that we can modify the binding of natural IgM to adenovirus. We introduced mutations that prevent virus inactivation in the bloodstream and its trapping in liver macrophages, the largest pool of immune cells in our body that trap and destroy pathogens,” he says.
“Up to now, the prevailing view has been that any regular repeating structure, like the shell of the virus, would attract low-affinity natural IgM antibody binding, leading to its prompt inactivation and removal from the blood.”
The researchers also replaced part of the adenovirus that interacts with human cellular integrins, substituting a sequence from another human protein, laminin-α1 that targets the virus to tumor cells. Emerson and Stewart obtained a high resolution cryo-electron microscopy structure of the re-engineered virus (see figures).
When injected into mice, high doses of standard adenovirus triggered liver damage and death within a few days, but the modified virus did not.
The modified virus could eliminate disseminated tumors from some, but not all mice engrafted with human lung cancer cells; a complete response – lack of detectable tumors and prolongation of survival – was observed in about thirty five percent of animals.
Tumor sites in the lung were converted into scar tissue, the scientists found. Now, Shayakhmetov’s lab is exploring approaches to further increase the proportion of complete responders.
In the clinic, metastatic lung cancer would be the type of cancer most appropriate to test an oncolytic virus against, Shayakhmetov says. The technology could also be harnessed for gene therapy applications.
It is a sad fact that most people who develop cancer die from it. The underpinnings of such drama lie in the fact that most cancers become incurable after their metastasis. A factor contributing to this end is the plasticity of tumor cell populations during the course of chemotherapy or radiotherapy, which often leads to tumor resistance.
At this juncture the therapeutic index narrows significantly rendering most available treatments ineffective and causing an insufferable stage in the life of a cancer patient, as a result of the high toxicity to normal tissues and to the patient as whole.
Except for some remarkable approaches involving the patient’s own immune system [1], there is no silver bullet to address the cancer problem, and the wiser approach where cancer could be treated as a chronic disease using non-toxic novel molecules, such as those in anti-angiogenic therapies, to keep it from progressing is emerging as a protracted war. However, even with their appealing modes of action, anti-angiogenic drugs produce only modest objective responses when administered as single agents [2, 3].
These agents typically are not able to enhance patients survival in clinical trials [4] and may need to be combined with chemotherapy to exert their therapeutic benefit [5, 6]. It was even suggested that an anti-angiogenic resistance mechanism could be developed by reducing tumor response to hypoxia through the loss of p53 function [7] or by switching pro-angiogenic factors [8].
There is thus an urgent need to diversify and to combine different strategies with non-overlapping anti-cancer modes of action to achieve a potentially successful anti-cancer therapy. Among the diversified arsenal of weapons against cancer is a particular class of agents that could bring much needed help: viruses.
It is ironic that we have spent the last few million years fighting these infectious agents by developing innate and active immunity against them and then turn to them, recently, to enlist them in the fight against cancer. The specific lysis of cancer cells following viral infection is an opportunistic event that favors viral cycle completion in cancer cells due to their propensity to progress through the S phase, which is often induced by viruses themselves.
The idea of using viruses as an anti-cancer drug was first proposed in 1904 when patients with malignancies who underwent viral infections or rabies vaccination were found to experience transient remissions [9, 10]. This finding led to a broad-range investigation identifying a large arsenal of novel oncolytic viruses with anti-tumor activity—38 viruses, including adenovirus, Bunyamwara, coxsackie, dengue, feline panleukemia, Ilheus, mumps Newcastle disease, vaccinia, and West Nile virus [11–14], which were tested in vivo in both animals and man.
Although all of these viruses replicate in cancer cells to a certain extent, none show the ideal attributes of a successful anti-cancer virus. T
hus they fail to
1) infect only cancer cells, due to the ubiquity of their receptors on normal as well as tumor cell surfaces,
2) they do not replicate specifically in cancer cells, because of the high constitutive promoter expression of viral genes necessary for viral replication, and
3) they are unable to avoid the detection and elimination by the immune system.
Positive attributes of adenoviruses include the findings that they do not cause serious human illnesses and have moderate side effects. Moreover, virus production can be safe and efficient to allow for the prospect of large-scale preparation and use.
Early studies using oncolytic viruses could not provide conclusive findings regarding the clinical utility of these agents. Indeed most of the studies used non-concentrated crude cell lysates, which limited the amount of virus to a suboptimal dose. The development of virology techniques and in particular of large-scale purification protocols allowed for subsequent more rigorous studies.
The extensive studies of potential oncolytic viruses in the years between 1950 and 1975 and in particular the landmark study performed by the National Cancer Institute [14] indicated the feasibility of using adenoviruses as oncolytic viruses for cancer treatment.
Sixty-five percent of the patients who were treated locally thus, showed moderate to marked local responses, translating into the ulceration and liquefaction of injected tumors, while no response was reported in patients whose tumors were injected with heat-inactivated adenoviruses. While the same study revealed that patients treated with replication-competent adenoviruses raised an immune response within 7 days after viral inoculation, viral particles were present in tumors even 17 days post-inoculation indicating viral replication in immune-competent hosts.
Other oncolytic viruses did not show such potential due mainly to their lack of selectivity and high toxicity [12]. Other compelling reasons to use adenoviruses for the purpose of gene therapy [15, 16] in general and cancer therapy in particular [17–19] are its dramatic transduction efficiency in vivo, its ease of preparation, use and safety profile, and the possibility of enhancing and modifying adenovirus tropism and oncolytic effect for specific applications in cancer therapy.
Moreover, since their first description in the early 1950’s [20], adenoviruses have been widely studied, and much is now known about the mechanism of cell entry and tropism as well as their replication cycle [21–26], making it possible to reroute their entry into specific cellular targets and to control the transcription of viral genes after viral infection.
In brief, we know a great deal more about adenoviruses than any other oncolytic virus. As a result such knowledge enables the ability to engineer safer and more useful conditionally replicating adenoviruses for the purpose of cancer gene therapy.
Adenoviral vectors and adenovirus-transduced cells are susceptible both to cytotoxic T-lymphocyte and humoral immune responses. Additionally, adenoviral-based vectors do not integrate their genome into the cellular, chromosomal DNA of transduced cell populations and therefore do not allow for long-term transgene expression.
For these reasons, adenoviral vectors are uniquely useful for genetic immunization programs against infectious diseases and for cancer therapy. The latter is the subject of this review.
ADENOVIRUSES
Members of the Adenovirus family are non-enveloped, non-integrating, lytic double-stranded DNA viruses with a genome ranging in size from 30–38 Kilobases and encoding a total of 30–40 genes “Fig. (1)”. Adenovirus infections occur primarily in children [27] and can infect a wide variety of well-differentiated dividing and resting cells, including liver, brain, lung, heart and skeletal muscle. Adenovirus infection in immunocompetent individuals is often mild and does not require medical treatment.
However, adenovirus infection could be life-threatening in immunocompromised individuals, such as AIDS patients, transplant recipients, and patients with hereditary immunodeficiency. The classification of adenoviruses is based mainly on immunological criteria such as serotypes[28].
To date, 51 human adenovirus serotypes have been categorized into 6 species: A to F. Adenovirus species differ in their usage of the adenovirus receptor [29–31] and show a preference for specific organs. For example, adenovirus species D infects the eyes, species A and F target the gastrointestinal tract while adenovirus species C, E, and some of members of species B typically infect the respiratory tract, while others from B species infect the urinary tract as well [32].

Adenovirus genome. The 30–38 Kb adenovirus genome is organized into multiple early (E) and late (L) regions of transcription. Initial induction of E1A within the E1 region leads to the downstream transcription and subsequent replication of the entire adenoviral genome. The majority of the late gene regions encode for structural virion elements (Figure 2) which are necessary for repackaging, lysis, and subsequent infection of other cells.
The 5’ ends of the adenovirus double-stranded genome are covalently attached to a terminal protein (TP) [33]. Inside the capsid the adenoviral DNA is wrapped around but non-covalently bound to the highly basic arginine-rich protein VII and the small (4 kDa) peptide mu [34] “Fig. (2)”.
An additional arginine-rich protein, V, is attached to the DNA and provides an anchor between the whole DNA-protein complex and the hexon of the capsid via yet another protein VI [35]. Adding to the complexity of this intricate structure is another crucial component, a virus encoded protease, which is needed to process the structural protein forming the capsid of mature viral particles [36, 37].
On the outside, adenoviruses have an icosahedral-shaped capsid of a particulate size of 80–100nm [38] and therefore are potentially capable of reaching tumor cells via tumor blood vessels’ leaky pores, which have an estimated diameter of 400–600nm [39].
The viral capsid consists of three major structural proteins: the hexon, the fiber, and the penton base, along with other minor proteins: VI, VIII, and IX, which are associated with the hexon, and IIIa, which is associated with the penton, and IVa2.

Adenovirus cell entry
Except for members of adenovirus species B, which recognizes and binds to a distinct receptor [40], adenoviruses bind to target cells by the knob of the fiber. This binding involves a high affinity interaction with the adenovirus receptor termed CAR (coxsackie/adenovirus receptor). CAR is a 46 kDa trans-membrane protein of the immunoglobulin superfamily [41], and is identical to coxsackie B virus receptor [42].
Additionally, adenovirus C (type 2 and type 5) can bind to the histocompatibility class I molecule, a member of the immunoglobulin superfamily [43]. Following receptor recognition, the penton base interacts, via its Arg-Gly-Asp (RGD) motif, with αv integrins [44, 45], especially αvβ3 and αvβ5 integrins.
These initial interactions are followed by the activation of signaling pathways that enforce adenovirus cellular entry of the adenovirus but, potentially, may simultaneously trigger the host’s immune system. The enforced entry signaling pathway is induced by the activation of phosphoinositide-3-OH kinase (PI-3K), which in turn activates the Rho family of GTPases and subsequently leads to the polymerization and reorganization of actin with the apparent goal of facilitating endocytosis [46, 47].
The other signaling pathway is responsible for the activation of the Raf/mitogen-activated protein kinase (MAPK), which is followed by IL-8 production as early as 20 min post-infection, potentially acting as a chemoattractant for leukocytes. Clathrin-coated endosomes containing the engulfed adenoviruses are then shuttled to the cytoplasm [48].
Inside the virus, the virus-encoded protease disrupts the association between the capsid and the complexed DNA/core proteins by proteolytic cleavage of the protein anchor VI [49]. After disruption of the capsid, the viral DNA is injected into the nucleus through a nuclear pore.
This passage to the nucleus requires the intervention of dynein and microtubules [50, 51]. One or two hours after infection, adenoviral DNA and proteins V and VII as well as viral particles can be detected in the nucleus [52, 53].
Replication of adenoviruses
Adenoviral replication events are quite common to all species and start by the active transcription of a battery of genes classified temporally as early and late genes. The early phase of replication is initiated by the transcription of several cassettes termed E1, E2, E3 and E4.
E1 transcripts are mainly designed to subdue cellular components that could hamper adenoviral replication and cycle completion. These transcripts are the E1A and E1B. E1A itself encodes two major proteins termed 32 kD and 26 kD “Fig. (1)”. E1A proteins are designed to modulate the functions of several major cellular proteins “Fig. (3)”, which have far reaching roles in cell division and cell fate. E1A proteins will bind to p21 and CDK inhibitors [54], cyclin A and E-CDK complexes [55], and p300/CBP transactivators thus interfering with transcriptional activities, modulating associated acetyltransferase activities associated with pCAF, and affecting the activity of STAT-1 which is required for interferon response and blocking caspase activation in p53-independent apoptosis [56–60]. E1A will also interfere with transcription by binding directly to the TATA-box-binding protein (TBP) and TBP-associated protein TAF [61].
Like T antigen (SV40), and E7 (HPV), E1A binds to and inactivates the retinoblastoma (Rb) tumor suppressor protein by using an E1A conserved LXCXE sequence motif [62]. The binding of E1A to Rb releases the transcription factor E2F which leads to the transcription of p53 thus promoting apoptosis [63] and of p19ARF which interacts with mdm2, thus preventing mdm2 from interacting with p53 [64] leading to a stabilization of p53 by avoiding its proteolysis by ubiquitination [65, 66].
The stabilization of p53 is further enhanced by the interaction of E1A with Sug1, a subunit of the proteasome complex that is required for p53 degradation [67]. All of these interactions are directed at subduing the transcriptional machinery of the infected cell to allow for adenovirus replication, and they seem to drive infected cells toward apoptosis since several interactions are aimed at enhancing p53 stability and activity. However, the second transcript group of the E1 region, E1B seems to counter this pro-apoptotic trend “Fig. (4)”.
The E1B product E1B-55 kDa seems to interact directly with p53 and mediates its inactivation [68] and translocation to the cytoplasm [69]. E1B-55 kDa does help mediate the ubiquitination and degradation of p53 in concert with another adenoviral protein E4orf6 [70, 71].
The interaction of E1B-55 kDa and p53 does in fact transform p53 into a very potent repressor while increasing p53 affinity to its binding site [72]. This observed relationship suggests that wild type adenoviruses reduce the tumor suppressor activity of p53. Furthermore, another product of the E1B gene, E1B-19 kDa, blocks the downstream effects of p53 to prevent apoptosis [73].
In this respect E1B-19 kDa functions as an analogous form of Bcl-2 and can inactivate the pro-apoptotic factor Bax [74]. To further antagonize the E1A proapoptotic functions, E1B-19 kDa can bind much like Bcl-2 to the powerful transcription repressor btf which promotes cell death by inducing mitochondrial membrane permeabilization [75, 76].
The anti-apoptotic function of E1B-19 kDa is truly revealed when deleted from the adenovirus genome itself. E1B-19 kDa deletion was shown to yield a highly oncolytic adenovirus [77], which lyses infected cells and spreads much faster than the wild type adenovirus. The deletion of both E1B products (E1B-55 kDa and E1B-19 kDa) in another adenovirus, termed Ad337, [78] resulted in similar increases in oncolytic effect and virus cytotoxicity.

E1A protein interaction network. E1A is the first protein translated from the viral genome. As such, it is responsible for the successful downstream transcription as well as replication of the entire viral genome in order to ensure viral infection and propagation.

E1B protein anti-apoptotic mechanisms. The two E1B region proteins, E1B-55 kDa and E1B-19 kDa, both result in apoptotic-prevention mechanisms to help ensure successful viral infection and spread. The E1B-55 kDa predominantly functions to sequester and inhibit p53 but can also indirectly act through E4orf6 to ubiquitinate and degrade p53 to prevent p53-activated apoptosis. The E1B-19 kDa protein acts as a Bcl-2 analog and inhibits Bax and/or Bak from signaling mitochondrial rupture, cytochrome c release, caspase activation, and ultimate apoptosis.
This interplay provides a counter measure to E1A activation of p53. A simplistic view of these complex interactions would seem to indicate that in order for adenoviruses to replicate they must first modulate the transcriptional machinery of the infected cell at the cost of triggering a pro-apoptotic program by the E1A gene product and then manage this crisis by the products of the E1B genes.
The E2 gene products are necessary for the replication of virus DNA and provide the tools for DNA replication and transcription in collaboration with other cellular components [79]. The transcription of the E2 genes is dependent on the E1 products, which act as trans-acting transcriptional factors.
The genes encoded by the E3 regions are not necessary to the replication of adenoviruses. However, the sophisticated function of some of its products is worth mentioning, in particular of the 19 kDa glycoprotein termed E3 gp19K. This protein is anchored to the ER and binds to the heavy chain of MHC class 1 but also delays its expression, thus preventing its shuttling to the cell surface for presentation and recognition by CTLs as well as an ultimate confrontation with the immune system [80].
Another protein termed adenovirus death protein (ADP) or E3-11.6K is known to promote cell lysis and the release of adenoviral particles [81]. Adenoviruses overexpressing ADP were found to induce early cell lysis, increased viral spread [82] and to provoke cell death by both caspase-dependent and independent mechanisms [83].
Due to the dispensable nature of these functions in relation to the replication of the adenovirus, the E3 region is commonly deleted from its genome to allow for the creation of valuable “real estate space” in order to incorporate large transgenes for the purpose of cancer gene therapy.
The quintessential example of an adenovirus with a deletion-modified E3 region is the famous Onyx-015 [84]. The E4 gene products main function is to shut-off cellular protein synthesis [85] and possibly cooperate with the E1B-55 kDa to allow the replication of the adenovirus in a cell cycle independent manner [86].
Finally, many transcripts are encoded toward the end of the virus cycle and are termed late genes L1 to L5, which result mostly in transcripts whose products correspond to the structural proteins of the capsid. Their transcription is delayed due to an attenuation of the major late promoter (MLP) in part because of fierce competition for transcriptional activators which are limited [87].
It is of interest to note that few adenoviral transcripts do not yield proteins. Among them are the Virus-associated (VA) RNAs which are noncoding, polymerase III-transcribed, 160-nucleotide single-stranded RNA molecules that fold into dsRNA and accumulate in the cytoplasm of adenovirus-infected cells [88] “Fig. (5)”.
For example, Adenovirus type 5 expresses two VA RNAs, VAI and VAII. Though dsRNAs usually activate an RNA-dependent protein kinase (PKR), an interferon-inducible serine-threonine protein kinase which leads to protein synthesis shut-off as a response to viral infection [89], the main function of adenoviral VAI RNAs is the inhibition of PKR activation [90].
A recent report suggested that VAI RNAs are processed to small RNAs and could behave as functional siRNAs or miRNAs to regulate viral components [91]. Interestingly, the Ras oncogene, which is overexpressed in many tumors is also able to inhibit PKR. This property was used to build an adenovirus without the viral associated RNA, thus enabling its replication only in Ras overexpressing cells [92].
Another report indicates that VA RNAs suppress RNA interference (RNAi) later after infection by suppressing the activity of two key enzyme, Dicer and RNA-induced silencing complex (RISC) [93].

Adenovirus VA RNA inhibition of both the Interferon/PKR pathway. VA RNAs (with imperfect-stemloop secondary structure) inhibit IFN-induced PKR signaling leading to downstream eIF2α phosphorylation and ultimate protein translation inhibition. Ras signaling via MEK and its substrate ERK may also inhibit IFN-induced PKR activation by preventing PKR phosphorylation. Additionally, VA RNAs may also be processed into siRNAs and inhibit the functionality of the host cell’s Dicer/RISC RNAi pathway, thus blocking degradation of short viral dsRNAs as well as the cell’s endogenous miRNA/siRNA synthesis pathway.
The assembly of the adenovirus is triggered by the encapsidation of viral DNA which contains an AT-rich packaging signal at its left end [94]. The exit of adenovirus particles from the nucleus where they matured is facilitated by the disruption of the nuclear membrane followed by the collapse of the plasma membrane without necessarily showing signs of apoptosis [81].
DESIGNING USEFUL ADENOVIRAL CONSTRUCTS FOR CANCER GENE THERAPY
Several properties of adenoviruses make them useful as vectors for cancer gene therapy. These include, the mild nature of illness resulting from adenovirus infections, the lack of integration into the host genome, their high transduction ability.
A wild type adenovirus has a “cargo” capacity of 2 kb for potential gene transfer, corresponding to ~ 105% of the original size of the genome [95].
This capacity is limited by the intra-capsid volume and steric interactions with the viral core proteins inside it. However, depending on the application, certain genes such as E1 and E3 may be deleted to increase this capacity to 7.5 kb [96]. A further increase in capacity to 11 kb may be achieved with an additional deletion in the E2 region [97].
For replication incompetent adenoviruses, also termed first generation adenoviruses, deletions of adenoviral genes to yield replication defective viruses are also an effort to make the virus less susceptible to generating wild type virion particles.
On one hand the deletion of the E1A genes will lead to a reduced transcription of the E2 genes, while deletion of E1B will enhance the pro-apoptotic signals in infected cells and finally the deletion of E3 will reduce the chances of virus-infected cells escaping immune responses [98, 99].
These deletions thus limit adenovirus usage in long-term gene therapy protocols but potentially enhance their application in vaccine development. Adenoviruses lacking E1 genes could be grown in a packaging cell line transformed with E1A and E1B genes [100].
This system provides viral preparation in excess of 1013 particles/ml, allowing for direct usage in in vivo applications [101]. Second generation replication-defective adenoviruses were later developed by excising some or all the adenoviral genes from the E2 and E4 regions [97, 102, 103].
However, eliminating all these adenoviral components does not abrogate the immune response to adenoviruses, as the basis of this response is not only triggered by the therapeutic transgene inserted into the adenoviral genome [104] but also by the required structural components of adenovirus [98, 105].
The following generations of adenovirus vectors were indeed aimed at removing the maximum number of genes from the genome to generate the so called “gutless” [106–108] vector. Gutless vectors contain ITRs and packaging signals but require a helper virus, which means that gutless adenoviruses needed special care for purification.
This problem was solved by incapacitating the packaging of the helper virus through the use of the cre-lox system [109]. Another variety of vectors emerged, called high capacity adenoviruses (HC-Ad) and in which most viral DNA was replaced by “stuffers” in order to permit efficient adenovirus packaging [106, 109–115].
HC-Ad are more suitable for therapeutic gene delivery of large genes and by contrast to their first generation predecessors were reported to allow for transgene expression for a surprisingly longer period of time, over one year [116].
ADENOVIRUSES FOR CANCER GENE THERAPY
The use of adenoviruses for cancer gene therapy involving the delivery of a therapeutic gene could be classified into three categories: 1) adenoviruses expressing tumor suppressor genes; 2) oncolytic adenoviruses potentially armed with prodrug-activating enzymes and; 3) adenoviruses for DNA vaccines. However, it is conceivable to unify all of these classes into a single one. For example, it is possible to design an oncolytic adenovirus which will be armed with a prodrug-activating enzyme and which could elicit an immune response. Another design could also combine tumor suppressor genes and immunomodulatory genes. A wide array of other combinations are described in the literature. However, in this review we will only discuss the first two categories.
Adenoviruses expressing tumor suppressor genes
Mutations or deletions leading to the loss of function of tumor suppressor genes and other genes involved in checkpoints controlling the quality of genetic material, cell division, survival and the death of cells, often lead to an imbalance of cell growth, which becomes unregulated, leading to an uncontrolled cell cycle. Such imbalance often leads to the development of cancer cells. The re-introduction of tumor suppressor genes or other genes involved in cellular growth control is thought to restore normal cellular functions [117].
Targeting the p53 pathway: anti-cancer chemotherapy utilizing 60,000 compounds against a panel of 60 human cancer cell lines demonstrated that it is most efficacious in tumor cell lines expressing a functional p53 gene [118]. However, most human cancers harbor a defective p53 gene [119]. Consequently, it was demonstrated that the introduction of a wild type p53 gene into p53 defective colorectal carcinoma leads to cell growth suppression [120]. A recent clinical trial using adenovirus-mediated p53 gene transfer in patients with chemo-radiation-resistant advanced esophageal carcinoma showed a clear anti-tumor effect [121]. Combining the transfer of p53 with chemotherapy is a good practice, which synergizes their actions. For example, it was found that the introduction of wild-type p53 in non-small lung cancer cells sensitizes them to cisplatin [122], and sensitizes myeloid leukemia cells to etoposide [123] and thyroid cancer cell lines to adriamycin [124]. Combination with radiotherapy was equally effective on radioresistant colon [125], ovarian [126], and radioresistant head and neck cancer cell lines in vivo [127]. However, the introduction of wild type p53 function or its abrogation does not enhance or reduce the radiosensitivity of some cell lines [128, 129], indicating a cell-type specific p53 radiosensitization. The overexpression of mouse double-minute 2 (mdm2) in certain cancers could limit the efficacy of p53 gene transfer, for example 30% of osteosarcoma tumors overexpress mdm2 due to gene amplification [130]. This was resolved through the creation of a chimeric p53 in which the domains that mediate its inactivation were replaced [131]. This chimeric p53 induced apoptosis six-fold more efficiently than p53 wild type. Other modifications to overcome the inactivation by mdm2 are the substitutions of hydrophobic residues Leu-14 and Phe-19 on p53, both of which are involved in the interaction with mdm2 [66]. The modified p53(14/19) was delivered by Ad-p53(14/19) and was found to induce apoptosis more dramatically than the wild type p53 especially in osteosarcoma cells overexpressing mdm2 [132].
Wild type p53 transfer will probably be ineffective in HPV induced cervical cancer since E6 protein of HPV inactivates p53 through ubiquitin-mediated proteolysis [133], in the SV40 large T-antigen expressing tumor cells in which the T-antigen inactivates p53 [134], or in hepatitis induced liver cancer where the X-protein excludes p53 from the nucleus [135].
Other tumor suppressor genes: approaches involving inhibition of cyclin-dependent kinases were explored by adenoviral delivery of p21 waf/cip1, a universal inhibitor of CDK’s and a mediator for p53 G1 arrest. p21 transfer induces apoptosis [136] and would be useful for those cancer cells overexpressing mdm2 resulting in inactivated p53. Indeed it was shown that p21 was more effective than p53 in a rat glioma [137]. In some cases however the lack of p21 is behind the enhanced sensitivity to chemotherapy [138, 139], thus throwing some doubt about the utility of p21 gene transfer.
p16INK4 deletions are encountered in over 50% of gliomas [140]. When comparing the impact of adenovirus transfer of p53, p21 and p16, it was found that while p16 induced a prostate cancer growth delay similar to p21 it was weaker than p53 [141]. However, surprisingly, the introduction of p16 into a melanoma cell line lead to a dramatic chemoresistance to methotraxate, vinblastine, and cisplatin [142]. Another tumor suppressor that could be considered is the Retinoblastoma (Rb) gene, which binds the E2F transcription factor when hypophosphorylated but releases it when hyperphosphorylated, thus allowing cell-cycle entry into S phase [143]. Rb gene deletions are present in lung, bladder, breast, and osteosarcoma and other cancers. Upon adenoviral transfer of the Rb gene to these cancers, Rb showed a hypophosphorylated status and lead to a complete tumor suppression of the bladder treated tumor cells in nude mice when using a N-terminal truncated retinoblastoma (RB) protein (pRB94) [144, 145].
p27Kip1 is another universal CDK inhibitor from the same family as p21 and mediates a similar growth arrest at G1 phase [146]. p27 was compared to p21 following adenovirus-mediated transfer in breast cancer and was found to decrease CDK activities more than p21 [147] and to inhibit tumor growth of glioma in vivo [148]. It appears however that the approach of delivering tumor suppressor genes using replication-defective adenoviruses is of limited utility since not all tumor cells will be successfully infected.
Among all the tumor suppressors discussed here, it appears that the use of the p53 mutants, resistant to mdm2 inactivation [131, 132] are the most likely candidates to proceed toward advanced clinical use. However, due to the limited spread imposed on replication-defective adenoviruses, it seems logical to deliver p53 using conditionally replicating adenoviruses (CRADs). There is, however, a potential problem with this concept. Since most E1 gene products are antagonists of p53, especially E1B-55 kDa which interacts directly with p53 and mediates its inactivation [68] and translocation to the cytoplasm [69]. Again, E1B-55 kDa also mediates the ubiquitination and degradation of p53 in concert with the adenoviral protein E4orf6 [70, 71]. The interaction of E1B-55 kDa and p53 does in fact transform p53 into a very potent repressor while increasing p53 affinity to its binding site [72]. This interaction suggests that E1B-55 kDa reduces the tumor suppressor activity of p53, which is the goal of p53 transfer in cancer cells in the first place. However, since E1B-55 kDa is produced early during the replication cycle of adenoviruses it seems sensible to place p53 gene under the control of a late promoter. This goal was achieved by placing p53 cDNA into the fiber transcription cassette [149]. The late production of p53 did not impair the cycle of the replication competent adenovirus. However, this elegant engineering did not mediate a strong p53 tumor suppressor function in this context despite its strong nuclear accumulation. The solution to this problem was provided by a p53 mutant in which the protein domain interacting with E1B-55 kDa was removed [150, 151] or by the deletion of the domain interacting with mdm2 such as the modified p53(14/19), which was delivered by Ad-p53(14/19) and was found to induce apoptosis more dramatically than the wild type p53 especially in osteosarcoma cells overexpressing mdm2 [132].
Tumor suppressor targeting strategies should provide a multimodal treatment involving 1) the tumor suppressor function while insuring that such property will be exacted in target tumor cells; 2) the additional oncolytic activity of CRAD to allow for the spread of the tumor suppressor gene to tumor cells; and 3) the possibility to combine the first approaches with chemotherapy or radiation.
Conditionally replicating adenoviruses (CRADs)
Like many DNA viruses, adenoviruses have developed intricate mechanisms to disrupt major cellular checkpoint effectors, such as p53, mdm2, and Rb, thus affecting several cellular pathways including cell cycle progression in a manner conducive to bringing the adenoviral cycle to completion.
Adenoviruses achieve this remarkable exploit by a very limited repertoire of genes whose products can achieve several protein-protein interactions via multiple domains within the same adenoviral protein “Fig. (3, ,4)”.4)”. However normal cells rather than tumor cells are the evolutionary target of adenoviruses. Therefore, adenovirus biology was first dictated and shaped to function in normal cells.
If the mechanisms used by wild-type adenoviruses to subvert the normal cells’ processes are altered by removal of specific genes from the adenovirus genome, then the replication of the modified adenovirus will be allowed only in cells with de facto subverted processes. It appears that in most tumor cells those adenoviral-targeted pathways are already defective thus giving the adenoviruses’ replication a green light signal.
CRADs generated by deletion of adenoviral genes
One of the first oncolytic adenoviruses that was developed for cancer therapy was Onyx-015 [84]. This adenovirus was built on a simple, yet very elegant premise. The wild type adenovirus after infecting a normal cell (say an airway, epithelium cell), will force cell entry into S phase via one of its early products, the E1A protein, which interacts with the Rb protein. Rb is usually closely associated with E2F transcription factors and this close association blocks cell cycle progression.
The interaction of E1A and Rb releases E2F transcription factors thus lifting the blockade and allows infected cells to progress into the S phase. By eliciting such progression, the replication machinery of the infected cell is activated and the adenovirus’ replication is a direct beneficiary [152].
The infected cell’s response is swift and tries to abort this hijacking by triggering the expression of p53 (see earlier) which should lead to apoptosis [153], thus eliminating the infected cell and preventing subsequent viral spread. This cellular response is, however, countered by another adenoviral protein E1B-55 kDa (see earlier), which inactivates p53 [154]. Onyx-015 is devoid of a functional E1B-55 kDa and is therefore “theoretically” unable to complete its cycle in cells possessing a functional p53. However, Onyx-015 will be able to do so if p53 is not functional, such as in the case of most human cancers [119].
The initial reports showed a greater sensitivity of cells lacking p53 [84]. The p53-based specificity of Onyx-015 was later challenged by the finding that cells possessing functional p53 were permissive to Onyx-015 [155]. Although, this finding could be counter-challenged by the possibility of an indirect deficiency in the p53 pathway due to for example the loss of p14ARF [156]; p14ARF down regulates mdm2, a ubiquitin ligase which degrades p53 (see earlier), thus allowing cancer cells with defective p14ARF or overexpressed mdm2 to behave as p53 defective cells even with a wild type p53 gene [157].
Moreover and probably more damaging to Onyx-015 theory, some tumor cell lines required E1B-55 kDa protein to allow the replication of Onyx-015 regardless of their p53 status [158]. We should remember that in addition to inactivating the tumor suppressor p53, E1B-55 kDa protein has two late functions including host protein synthesis shut-off and the transport of late adenoviral mRNA [159, 160].
It was later found that the late mRNA transport is allowed in some tumor cell lines but not others and only where the export was allowed was Onyx-015 replication permissible, thus defining the true mechanism behind 0nyx-015 selectivity [161]. O’Shea and colleagues also showed that non-permissive cells would become permissive after a heat shock which allows late mRNA transport thus pheno-copying of E1B-55 kDa functions [162].
The anti-tumor mechanism of Onyx-015 is therefore not universal. Other oncolytic adenoviruses were generated by deleting both E1B-55 kDa and E1B-19 kDa for p53 and Rb null cancers [78, 163], by deletion of VA-RNA for Ras positive cancers [92], or by deleting E1B-55 kDa and modifying the E1A/Rb interaction domain in the constant region CR2 [164, 165] (Table 1).
Table 1
Oncolytic Adenoviruses. Some viruses gain tumor-replication selectivity based upon novel gene deletions within their viral genome; others gain selectivity based upon cancer-associated, specific promoter induction.
| Adenoviruses | Tumor-specificity | Clinical Trial | Ref |
|---|---|---|---|
| Wild type | None | N/A | [14, 236] |
| dl1520 (ONYX-015) | E1B-55 kDa-deletion/p53 inhibition | Phases I–III | [84] |
| dl337 | None | N/A | [237] |
| dl118 | E1B-deletion/p53 inhibition | N/A | [238] |
| hTERT-Ad | hTERT promoter specific induction | N/A | [239] |
| ONYX-411 | E2F-1 promoter | N/A | [240] |
| AD.DF3-E1 | DF3 promoter | N/A | [167] |
| CV787 | Rat probasin & PSA promoters | Phase I | [241] |
| CV764 | Human glandular kallikrein & PSA promoters | N/A | [242] |
| CN706 | PSA promoter | Phase I | [19] |
DMINISTRATION OF ONCOLYTIC ADENOVIRUSES FOR CANCER THERAPY
Onyx-015 was the first conditionally-replicating oncolytic adenovirus to be tested in a clinical trial. The safety and anti-tumor efficacy of this type of oncolytic adenoviruses is now evident. The therapeutic efficiency of oncolytic adenoviruses will be dependent on the route of administration. Intratumoral administration is by far the most common modality for administering these agents.
This modality increases targeting and enhances the chances of the initial tumor cells’ infection. Intratumoral injection would require far less adenovirus than if injected intravenously, intra-arterial or intraperitoneally. These last three modalities require an enormous amount of adenoviral particles: ~2×1013 pfu.
This enormous load could trigger an immune response and is often behind the high frequency of flu-like symptoms observed for patients receiving the oncolytic adenoviruses via these routes.
Other symptoms accompanying adenovirus injections are the elevation of liver transaminases, hyperbilirubnemia, and increases in inflammatory cytokines (TNFα) and interleukins IL-1 and 6 as well as interferon γ [193, 196].
The tragic death of Jesse Gelsinger 7 years ago during a clinical trial at the University of Pennsylvania in which he received 4×1013 pfu for the treatment of ornithine decarboxylase deficiency [216] shows the need to generate adenoviruses with reduced immunogenicity and higher oncolytic effect. Intratumoral injection was 1000-fold more effective than systemic injections in tumor xenografts [217].
While intratumoral injection would be ideal for localized solid tumors, for metastatic tumors a systemic injection would be required in which case the presence of pre-existing neutralizing antibodies will be an impediment to the success of this strategy and a combination with another treatment such as chemotherapy would be advisable.
This problem is compounded by the absence of an animal model and therefore only studies of human patients in clinical trials will help understand how to solve this problem.
reference link : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3354698/
More information: S. Atasheva el al., “Systemic cancer therapy with engineered adenovirus that evades innate immunity,” Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/ … scitranslmed.abc6659



{kind=link}