










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Molti ricercatori sul cancro possono affermare di aver ideato “bombe intelligenti”.
Quello che è mancato è il bombardiere invisibile, un sistema di consegna che può scivolare attraverso le difese radar del corpo.
I virus oncolitici, o virus che uccidono preferenzialmente le cellule tumorali, sono stati discussi e testati per decenni.
Un virus oncolitico contro il melanoma è stato approvato dalla FDA nel 2015. Ma contro i tumori metastatici, hanno sempre affrontato una barriera schiacciante: il sistema immunitario umano, che cattura rapidamente i virus iniettati nel sangue e li invia al fegato, la spazzatura del corpo disposizione.
I ricercatori di Emory e Case Western Reserve hanno ora aggirato quella barriera. Hanno riprogettato l’adenovirus umano, in modo che il virus non venga facilmente catturato da parti del sistema immunitario innato.
Ciò consente di iniettare il virus nel sangue, senza provocare una massiccia reazione infiammatoria.
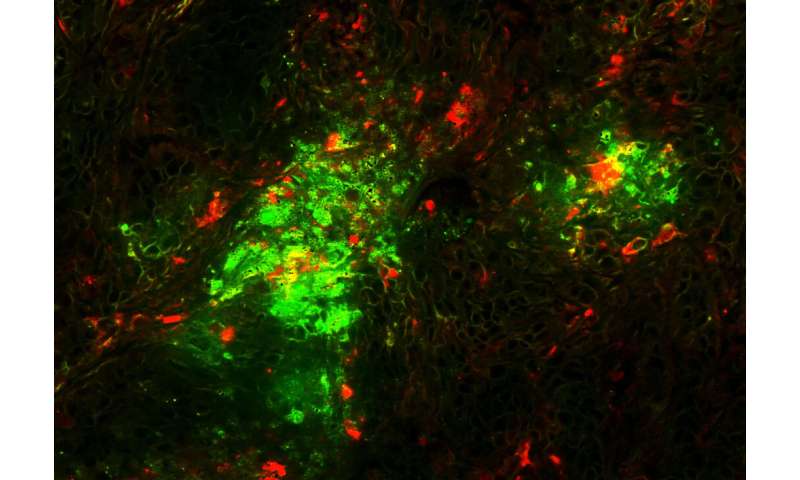
Una struttura di microscopia crioelettronica del virus riprogettato e la capacità del virus di eliminare i tumori disseminati nei topi sono riportati in Science Translational Medicine.
“Il sistema immunitario innato è abbastanza efficiente nell’inviare virus al fegato quando vengono somministrati per via endovenosa”, afferma l’autore principale Dmitry Shayakhmetov, Ph.D.
“Per questo motivo, la maggior parte dei virus oncolitici viene rilasciata direttamente nel tumore, senza influenzare le metastasi. Al contrario, pensiamo che sarà possibile somministrare il nostro virus modificato a livello sistemico a dosi sufficientemente elevate da sopprimere la crescita del tumore, senza innescare tossicità sistemiche potenzialmente letali “.
Adenovirus ingegnerizzato Ad5-3M con mutazioni evidenziate in rosso che sono state introdotte per indirizzare il virus alle cellule tumorali, ridurre l’infiammazione ed evitare interazioni con fattori del sangue e cellule immunitarie dopo la somministrazione sistemica Credito: Dmitry Shayakhmetov
I co-primi autori del documento Science Translational Medicine sono la scienziata associata di Emory Svetlana Atasheva, Ph.D. e Corey Emerson, studente laureato della Case Western Reserve. Shayakhmetov è professore di medicina e pediatria alla Emory University School of Medicine e membro del Lowance Center for Human Immunology and Emory Vaccine Center.
Shayakhmetov lavora da 15 anni con la biologa strutturale Phoebe Stewart, Ph.D., professoressa presso il Dipartimento di Farmacologia e membro del Cleveland Center for Membrane and Structural Biology presso la Case Western Reserve University. Il loro obiettivo: reingegnerizzare l’ adenovirus , un sistema di somministrazione che è stato utilizzato in dozzine di studi clinici sul cancro per stimolare la risposta antitumorale dell’ospite.
Gli adenovirus sono stati anche centrali negli studi sulla terapia genica. Shayakhmetov ricorda la morte nel 1999 di Jesse Gelsinger, un volontario in uno studio clinico di terapia genica morto per tempesta di citochine e insufficienza multiorgano connessa con alte dosi di un vettore di adenovirus immesso nel flusso sanguigno.
Dice che quell’evento lo ha ispirato a riorganizzare l’adenovirus, in modo che non scatenasse una forte reazione infiammatoria.
Egli vede l’ adenovirus riprogettato come una piattaforma tecnologica, che può essere adattata e personalizzata per molti tipi di cancro, e anche per i singoli malati di cancro come una forma di terapia personalizzata contro il cancro.
“Questa è una nuova strada per il trattamento dei tumori metastatici”, dice Shayakhmetov.
“Puoi armarlo con geni e proteine che stimolano l’immunità al cancro e puoi assemblare il capside, un guscio del virus, come se stessi mettendo dei blocchi Lego”.

Shayakhmetov ha iniziato a lavorare sulla tecnologia dei virus modificati mentre era all’Università di Washington e ha fondato una società, AdCure Bio, per portare una terapia potenzialmente salvavita ai pazienti con malattia metastatica.
Nel 2012, i laboratori di Shayakhmetov e Stewart hanno pubblicato un’analisi crio-EM su come l’adenovirus interagisce con un fattore ospite nel sangue, il fattore X della coagulazione, su Science.
“A volte anche piccoli cambiamenti nelle proteine strutturali possono essere catastrofici e impedire l’assemblaggio del virus infettivo”, afferma Stewart.
“In questo caso, abbiamo modificato l’adenovirus in tre punti per ridurre al minimo le interazioni del virus con specifici fattori del sangue. Abbiamo scoperto che il virus si assembla e rimane funzionale per infettare e uccidere le cellule tumorali “.
È ancora possibile che una risposta immunitaria adattativa a costruzione più lenta si sviluppi al virus modificato, simile a quella osservata con un vaccino. Un gruppo di virus potrebbe essere utilizzato per la somministrazione sequenziale ai malati di cancro per estendere i benefici terapeutici, dice Shayakhmetov.
“Il nostro studio è il primo a dimostrare che possiamo modificare il legame delle IgM naturali all’adenovirus. Abbiamo introdotto mutazioni che impediscono l’inattivazione del virus nel flusso sanguigno e il suo intrappolamento nei macrofagi del fegato, il più grande pool di cellule immunitarie nel nostro corpo che intrappolano e distruggono gli agenti patogeni “, afferma.
“Fino ad ora, l’opinione prevalente era che qualsiasi struttura ripetitiva regolare, come il guscio del virus, attrarrebbe il legame naturale degli anticorpi IgM a bassa affinità, portando alla sua pronta inattivazione e rimozione dal sangue”.
I ricercatori hanno anche sostituito parte dell’adenovirus che interagisce con le integrine cellulari umane, sostituendo una sequenza di un’altra proteina umana, la laminina-α1, che indirizza il virus alle cellule tumorali. Emerson e Stewart hanno ottenuto una struttura di microscopia crioelettronica ad alta risoluzione del virus riprogettato (vedi figure).
Quando iniettate nei topi, alte dosi di adenovirus standard hanno provocato danni al fegato e morte entro pochi giorni, ma il virus modificato no.
Il virus modificato potrebbe eliminare i tumori disseminati da alcuni, ma non da tutti i topi innestati con cellule di cancro del polmone umano; una risposta completa – mancanza di tumori rilevabili e prolungamento della sopravvivenza – è stata osservata in circa il trentacinque per cento degli animali.
I siti tumorali nel polmone sono stati convertiti in tessuto cicatriziale, hanno scoperto gli scienziati. Ora, il laboratorio di Shayakhmetov sta esplorando approcci per aumentare ulteriormente la percentuale di rispondenti completi.
Nella clinica, il cancro del polmone metastatico sarebbe il tipo di cancro più appropriato per testare un virus oncolitico, dice Shayakhmetov. La tecnologia potrebbe anche essere utilizzata per applicazioni di terapia genica.
È un fatto triste che la maggior parte delle persone che sviluppano il cancro muoiano a causa di esso. Le basi di tale dramma risiedono nel fatto che la maggior parte dei tumori diventa incurabile dopo le loro metastasi. Un fattore che contribuisce a questo scopo è la plasticità delle popolazioni di cellule tumorali durante il corso della chemioterapia o della radioterapia, che spesso porta alla resistenza del tumore.
In questo frangente l’indice terapeutico si restringe notevolmente rendendo inefficaci la maggior parte dei trattamenti disponibili e determinando una fase insopportabile nella vita di un malato di cancro, a causa dell’elevata tossicità per i tessuti normali e per il paziente nel suo insieme.
Fatta eccezione per alcuni notevoli approcci che coinvolgono il sistema immunitario del paziente [1], non esiste un proiettile d’argento per affrontare il problema del cancro e l’approccio più saggio in cui il cancro potrebbe essere trattato come una malattia cronica utilizzando nuove molecole non tossiche, come quelle in terapie anti-angiogeniche, per evitare che progredisca sta emergendo come una guerra prolungata. Tuttavia, anche con le loro attraenti modalità di azione, i farmaci anti-angiogenici producono solo modeste risposte oggettive quando somministrati come agenti singoli [2, 3].
Questi agenti tipicamente non sono in grado di aumentare la sopravvivenza dei pazienti negli studi clinici [4] e potrebbe essere necessario combinarli con la chemioterapia per esercitare il loro beneficio terapeutico [5, 6]. È stato anche suggerito che un meccanismo di resistenza anti-angiogenica potrebbe essere sviluppato riducendo la risposta del tumore all’ipossia attraverso la perdita della funzione di p53 [7] o cambiando i fattori pro-angiogenici [8].
Vi è quindi un’urgente necessità di diversificare e di combinare diverse strategie con modalità di azione anti-cancro non sovrapposte per ottenere una terapia anti-cancro potenzialmente efficace. Tra l’arsenale diversificato di armi contro il cancro c’è una particolare classe di agenti che potrebbero portare l’aiuto tanto necessario: i virus.
È ironico che abbiamo passato gli ultimi milioni di anni a combattere questi agenti infettivi sviluppando un’immunità innata e attiva contro di loro e poi ci siamo rivolti a loro, di recente, per arruolarli nella lotta contro il cancro. La lisi specifica delle cellule tumorali a seguito di infezione virale è un evento opportunistico che favorisce il completamento del ciclo virale nelle cellule cancerose a causa della loro propensione a progredire attraverso la fase S, spesso indotta dai virus stessi.
L’idea di utilizzare i virus come farmaco antitumorale fu proposta per la prima volta nel 1904, quando si scoprì che pazienti con tumori maligni sottoposti a infezioni virali o vaccinazione antirabbica sperimentavano remissioni transitorie [9, 10]. Questa scoperta ha portato a un’indagine ad ampio raggio che ha identificato un ampio arsenale di nuovi virus oncolitici con attività antitumorale: 38 virus, tra cui adenovirus, Bunyamwara, coxsackie, dengue, panleucemia felina, Ilheus, parotite malattia di Newcastle, vaccinia e virus del Nilo occidentale [11-14], che sono stati testati in vivo sia sugli animali che sull’uomo.
Sebbene tutti questi virus si replichino in una certa misura nelle cellule tumorali, nessuno mostra gli attributi ideali di un virus anti-cancro di successo. T
ma non ci riescono
1) infettare solo le cellule tumorali, a causa dell’ubiquità dei loro recettori sulla superficie delle cellule normali e tumorali,
2) non si replicano specificamente nelle cellule tumorali, a causa dell’elevata espressione del promotore costitutivo dei geni virali necessari per la replicazione virale, e
3) non sono in grado di evitare il rilevamento e l’eliminazione da parte del sistema immunitario.
Gli attributi positivi degli adenovirus includono i risultati che non causano gravi malattie umane e hanno effetti collaterali moderati. Inoltre, la produzione di virus può essere sicura ed efficiente per consentire la prospettiva di una preparazione e un utilizzo su larga scala.
I primi studi che utilizzano virus oncolitici non sono stati in grado di fornire risultati conclusivi sull’utilità clinica di questi agenti. In effetti, la maggior parte degli studi utilizzava lisati cellulari grezzi non concentrati, che limitavano la quantità di virus a una dose non ottimale. Lo sviluppo di tecniche virologiche ed in particolare di protocolli di purificazione su larga scala ha consentito successivi studi più rigorosi.
Gli ampi studi sui potenziali virus oncolitici negli anni tra il 1950 e il 1975 e in particolare lo studio di riferimento svolto dal National Cancer Institute [14] hanno indicato la fattibilità dell’uso degli adenovirus come virus oncolitici per il trattamento del cancro.
Il sessantacinque per cento dei pazienti che sono stati trattati localmente in questo modo, ha mostrato risposte locali da moderate a marcate, che si sono tradotte in ulcerazione e liquefazione dei tumori iniettati, mentre nessuna risposta è stata riportata in pazienti i cui tumori sono stati iniettati con adenovirus inattivati dal calore. Mentre lo stesso studio ha rivelato che i pazienti trattati con adenovirus competenti per la replicazione hanno aumentato una risposta immunitaria entro 7 giorni dall’inoculazione virale, particelle virali erano presenti nei tumori anche 17 giorni dopo l’inoculazione, indicando la replicazione virale in ospiti immunocompetenti.
Altri virus oncolitici non hanno mostrato tale potenziale a causa principalmente della loro mancanza di selettività e alta tossicità [12]. Altri validi motivi per utilizzare gli adenovirus ai fini della terapia genica [15, 16] in generale e della terapia del cancro in particolare [17-19] sono la sua straordinaria efficienza di trasduzione in vivo, la sua facilità di preparazione, utilizzo e profilo di sicurezza e la possibilità di migliorare e modificare il trofismo dell’adenovirus e l’effetto oncolitico per applicazioni specifiche nella terapia del cancro.
Inoltre, sin dalla loro prima descrizione nei primi anni ’50 [20], gli adenovirus sono stati ampiamente studiati, e molto si sa ora sul meccanismo dell’ingresso cellulare e del tropismo, nonché sul loro ciclo di replicazione [21-26], rendendo possibile il reindirizzamento il loro ingresso in specifici target cellulari e per controllare la trascrizione di geni virali dopo infezione virale.
In breve, sappiamo molto di più sugli adenovirus di qualsiasi altro virus oncolitico. Di conseguenza, tale conoscenza consente la capacità di progettare adenovirus di replica condizionale più sicuri e più utili ai fini della terapia genica del cancro.
I vettori adenovirali e le cellule trasdotte da adenovirus sono suscettibili sia ai linfociti T citotossici che alle risposte immunitarie umorali. Inoltre, i vettori adenovirali non integrano il loro genoma nel DNA cellulare cromosomico di popolazioni cellulari trasdotte e quindi non consentono l’espressione del transgene a lungo termine.
Per questi motivi, i vettori adenovirali sono particolarmente utili per i programmi di immunizzazione genetica contro le malattie infettive e per la terapia del cancro. Quest’ultimo è l’oggetto di questa recensione.
ADENOVIRUS
I membri della famiglia degli Adenovirus sono virus litici a DNA a doppia elica senza involucro, non integranti, con un genoma di dimensioni comprese tra 30-38 kilobasi e codificante per un totale di 30-40 geni “Fig. (1) “. Le infezioni da adenovirus si verificano principalmente nei bambini [27] e possono infettare un’ampia varietà di cellule in divisione e a riposo ben differenziate, tra cui fegato, cervello, polmone, cuore e muscoli scheletrici. L’infezione da adenovirus negli individui immunocompetenti è spesso lieve e non richiede cure mediche.
Tuttavia, l’infezione da adenovirus potrebbe essere pericolosa per la vita in individui immunocompromessi, come pazienti affetti da AIDS, trapiantati e pazienti con immunodeficienza ereditaria. La classificazione degli adenovirus si basa principalmente su criteri immunologici come i sierotipi [28].
Ad oggi, 51 sierotipi di adenovirus umano sono stati classificati in 6 specie: da A a F. Le specie di adenovirus differiscono nel loro utilizzo del recettore dell’adenovirus [29–31] e mostrano una preferenza per organi specifici. Ad esempio, le specie di adenovirus D infettano gli occhi, le specie A e F prendono di mira il tratto gastrointestinale mentre le specie di adenovirus C, E e alcuni membri della specie B tipicamente infettano le vie respiratorie, mentre altre della specie B infettano anche le vie urinarie [ 32].

Genoma dell’adenovirus. Il genoma dell’adenovirus di 30-38 Kb è organizzato in più regioni di trascrizione precoce (E) e tardiva (L). L’induzione iniziale di E1A all’interno della regione E1 porta alla trascrizione a valle e alla successiva replicazione dell’intero genoma adenovirale. La maggior parte delle regioni del gene tardivo codifica per elementi virionici strutturali (Figura 2) che sono necessari per il riconfezionamento, la lisi e la successiva infezione di altre cellule.
Le estremità 5 ‘del genoma a doppio filamento dell’adenovirus sono attaccate in modo covalente a una proteina terminale (TP) [33]. All’interno del capside il DNA adenovirale è avvolto ma legato in modo non covalente alla proteina VII ricca di arginina altamente basica e al piccolo peptide mu (4 kDa) [34] “Fig. (2) “.
Un’ulteriore proteina ricca di arginina, V, è attaccata al DNA e fornisce un ancoraggio tra l’intero complesso DNA-proteina e l’esone del capside tramite un’altra proteina VI [35]. Alla complessità di questa struttura intricata si aggiunge un altro componente cruciale, una proteasi codificata da virus, necessaria per processare la proteina strutturale che forma il capside delle particelle virali mature [36, 37].
All’esterno, gli adenovirus hanno un capside di forma icosaedrica con una dimensione del particolato di 80-100 nm [38] e quindi sono potenzialmente in grado di raggiungere le cellule tumorali attraverso i pori permeabili dei vasi sanguigni del tumore, che hanno un diametro stimato di 400-600 nm [39 ].
Il capside virale è costituito da tre principali proteine strutturali: l’esone, la fibra e la base del pentone, insieme ad altre proteine minori: VI, VIII e IX, che sono associate all’esone, e IIIa, che è associata al pentone e IVa2.

Ingresso nelle cellule di adenovirus
Fatta eccezione per i membri dell’adenovirus specie B, che riconosce e si lega a un recettore distinto [40], gli adenovirus si legano alle cellule bersaglio tramite la manopola della fibra. Questo legame implica un’interazione ad alta affinità con il recettore dell’adenovirus chiamato CAR (recettore coxsackie / adenovirus). CAR è una proteina trans-membrana da 46 kDa della superfamiglia delle immunoglobuline [41] ed è identica al recettore del virus coxsackie B [42].
Inoltre, l’adenovirus C (tipo 2 e tipo 5) può legarsi alla molecola di istocompatibilità di classe I, un membro della superfamiglia delle immunoglobuline [43]. Dopo il riconoscimento del recettore, la base del pentone interagisce, tramite il suo motivo Arg-Gly-Asp (RGD), con le integrine αv [44, 45], in particolare le integrine αvβ3 e αvβ5.
Queste interazioni iniziali sono seguite dall’attivazione di vie di segnalazione che impongono l’ingresso cellulare dell’adenovirus dell’adenovirus ma, potenzialmente, possono contemporaneamente attivare il sistema immunitario dell’ospite. La via di segnalazione forzata all’ingresso è indotta dall’attivazione della fosfoinositide-3-OH chinasi (PI-3K), che a sua volta attiva la famiglia Rho di GTPasi e successivamente porta alla polimerizzazione e riorganizzazione dell’actina con l’apparente obiettivo di facilitare l’endocitosi [ 46, 47].
L’altra via di segnalazione è responsabile dell’attivazione della proteina chinasi attivata da Raf / mitogeno (MAPK), che è seguita dalla produzione di IL-8 già 20 minuti dopo l’infezione, che potenzialmente agisce come un chemiotattico per i leucociti. Gli endosomi rivestiti di clatrina contenenti gli adenovirus inghiottiti vengono quindi inviati al citoplasma [48].
All’interno del virus, la proteasi codificata dal virus interrompe l’associazione tra il capside e le proteine DNA / core complessate mediante la scissione proteolitica dell’ancora proteica VI [49]. Dopo la rottura del capside, il DNA virale viene iniettato nel nucleo attraverso un poro nucleare.
Questo passaggio al nucleo richiede l’intervento di dineina e microtubuli [50, 51]. Una o due ore dopo l’infezione, nel nucleo possono essere rilevati DNA adenovirale e proteine V e VII nonché particelle virali [52, 53].
Replicazione di adenovirus
Gli eventi di replicazione adenovirale sono abbastanza comuni a tutte le specie e iniziano dalla trascrizione attiva di una batteria di geni classificati temporalmente come geni precoci e tardivi. La fase iniziale della replicazione è iniziata dalla trascrizione di diverse cassette denominate E1, E2, E3 ed E4.
I trascritti E1 sono progettati principalmente per sottomettere i componenti cellulari che potrebbero ostacolare la replicazione adenovirale e il completamento del ciclo. Queste trascrizioni sono E1A e E1B. Lo stesso E1A codifica per due principali proteine chiamate 32 kD e 26 kD “Fig. (1) “. Le proteine E1A sono progettate per modulare le funzioni di molte delle principali proteine cellulari “Fig. (3) “, che hanno ruoli di vasta portata nella divisione cellulare e nel destino cellulare. Le proteine E1A si legheranno agli inibitori di p21 e CDK [54], ai complessi della ciclina A ed E-CDK [55] e ai transattivatori di p300 / CBP, interferendo così con le attività trascrizionali, modulando le attività dell’acetiltransferasi associate associate al pCAF e influenzando l’attività di STAT- 1 necessario per la risposta all’interferone e il blocco dell’attivazione della caspasi nell’apoptosi indipendente da p53 [56-60].
Come l’antigene T (SV40) e E7 (HPV), E1A si lega e inattiva la proteina soppressore tumorale del retinoblastoma (Rb) utilizzando un motivo di sequenza LXCXE conservato E1A [62]. Il legame di E1A a Rb rilascia il fattore di trascrizione E2F che porta alla trascrizione di p53 promuovendo così l’apoptosi [63] e di p19ARF che interagisce con mdm2, impedendo così a mdm2 di interagire con p53 [64] portando a una stabilizzazione di p53 evitando la sua proteolisi per ubiquitinazione [65, 66].
La stabilizzazione di p53 è ulteriormente migliorata dall’interazione di E1A con Sug1, una subunità del complesso proteasoma necessaria per la degradazione di p53 [67]. Tutte queste interazioni sono dirette a sottomettere il meccanismo trascrizionale della cellula infetta per consentire la replicazione dell’adenovirus e sembrano guidare le cellule infette verso l’apoptosi poiché diverse interazioni mirano a migliorare la stabilità e l’attività di p53. Tuttavia, il secondo gruppo di trascrizioni della regione E1, E1B, sembra contrastare questa tendenza pro-apoptotica “Fig. (4) “.
Il prodotto E1B E1B-55 kDa sembra interagire direttamente con p53 e media la sua inattivazione [68] e traslocazione nel citoplasma [69]. E1B-55 kDa aiuta a mediare l’ubiquitinazione e la degradazione di p53 insieme a un’altra proteina adenovirale E4orf6 [70, 71].
L’interazione di E1B-55 kDa e p53 infatti trasforma p53 in un potente repressore aumentando l’affinità di p53 al suo sito di legame [72]. Questa relazione osservata suggerisce che gli adenovirus wild type riducono l’attività oncosoppressore di p53. Inoltre, un altro prodotto del gene E1B, E1B-19 kDa, blocca gli effetti a valle di p53 per prevenire l’apoptosi [73].
A questo proposito E1B-19 kDa funziona come una forma analoga di Bcl-2 e può inattivare il fattore pro-apoptotico Bax [74]. Per antagonizzare ulteriormente le funzioni proapoptotiche E1A, E1B-19 kDa può legarsi molto come Bcl-2 al potente repressore della trascrizione btf che promuove la morte cellulare inducendo la permeabilizzazione della membrana mitocondriale [75, 76].
La funzione anti-apoptotica di E1B-19 kDa è veramente rivelata quando viene eliminata dal genoma stesso dell’adenovirus. È stato dimostrato che la delezione di E1B-19 kDa produce un adenovirus altamente oncolitico [77], che lisi le cellule infette e si diffonde molto più velocemente dell’adenovirus wild type. La delezione di entrambi i prodotti E1B (E1B-55 kDa e E1B-19 kDa) in un altro adenovirus, denominato Ad337, [78] ha determinato aumenti simili nell’effetto oncolitico e nella citotossicità del virus.

Rete di interazione della proteina E1A. E1A è la prima proteina tradotta dal genoma virale. In quanto tale, è responsabile del successo della trascrizione a valle e della replicazione dell’intero genoma virale al fine di garantire l’infezione e la propagazione virale.

Meccanismi anti-apoptotici della proteina E1B. Le due proteine della regione E1B, E1B-55 kDa ed E1B-19 kDa, risultano entrambe in meccanismi di prevenzione dell’apoptosi per aiutare a garantire il successo dell’infezione virale e la diffusione. L’E1B-55 kDa funziona prevalentemente per sequestrare e inibire la p53, ma può anche agire indirettamente attraverso E4orf6 per ubiquitinare e degradare la p53 per prevenire l’apoptosi attivata da p53. La proteina E1B-19 kDa agisce come un analogo di Bcl-2 e inibisce Bax e / o Bak dalla segnalazione di rottura mitocondriale, rilascio del citocromo c, attivazione della caspasi e apoptosi finale.
Questa interazione fornisce una contromisura all’attivazione E1A di p53. Una visione semplicistica di queste complesse interazioni sembrerebbe indicare che per replicare gli adenovirus devono prima modulare il meccanismo trascrizionale della cellula infetta al costo di innescare un programma pro-apoptotico da parte del prodotto del gene E1A e quindi gestire questa crisi mediante i prodotti dei geni E1B.
I prodotti del gene E2 sono necessari per la replicazione del DNA del virus e forniscono gli strumenti per la replicazione e la trascrizione del DNA in collaborazione con altri componenti cellulari [79]. La trascrizione dei geni E2 dipende dai prodotti E1, che agiscono come fattori trascrizionali trans-attivi.
I geni codificati dalle regioni E3 non sono necessari per la replicazione degli adenovirus. Tuttavia, vale la pena menzionare la sofisticata funzione di alcuni dei suoi prodotti, in particolare della glicoproteina da 19 kDa denominata E3 gp19K. Questa proteina è ancorata all’ER e si lega alla catena pesante dell’MHC di classe 1, ma ne ritarda anche l’espressione, impedendone così il passaggio alla superficie cellulare per la presentazione e il riconoscimento da parte dei CTL, nonché un confronto finale con il sistema immunitario [80] .
Un’altra proteina chiamata proteina della morte dell’adenovirus (ADP) o E3-11.6K è nota per promuovere la lisi cellulare e il rilascio di particelle adenovirali [81]. È stato scoperto che gli adenovirus che sovraesprimono ADP inducono la lisi cellulare precoce, una maggiore diffusione virale [82] e provocano la morte cellulare sia per meccanismi indipendenti dalla caspasi [83].
A causa della natura superflua di queste funzioni in relazione alla replicazione dell’adenovirus, la regione E3 viene comunemente eliminata dal suo genoma per consentire la creazione di un prezioso “spazio immobiliare” al fine di incorporare grandi transgeni ai fini del gene del cancro terapia.
L’esempio per eccellenza di un adenovirus con una regione E3 modificata dalla delezione è il famoso Onyx-015 [84]. La funzione principale dei prodotti del gene E4 è quella di interrompere la sintesi proteica cellulare [85] ed eventualmente cooperare con l’E1B-55 kDa per consentire la replicazione dell’adenovirus in modo indipendente dal ciclo cellulare [86].
Infine, molte trascrizioni sono codificate verso la fine del ciclo del virus e sono chiamate geni tardivi da L1 a L5, che risultano principalmente in trascrizioni i cui prodotti corrispondono alle proteine strutturali del capside. La loro trascrizione è ritardata a causa di un’attenuazione del principale promotore tardivo (MLP) in parte a causa della feroce competizione per gli attivatori trascrizionali che sono limitati [87].
È interessante notare che pochi trascritti adenovirali non producono proteine. Tra questi ci sono gli RNA associati al virus (VA) che sono molecole di RNA a filamento singolo non codificanti, trascritto dalla polimerasi III, 160 nucleotidi che si piegano in dsRNA e si accumulano nel citoplasma delle cellule infettate da adenovirus [88] “Fig. (5) “.
Ad esempio, l’Adenovirus di tipo 5 esprime due VA RNA, VAI e VAII. Sebbene i dsRNA di solito attivino una protein chinasi RNA-dipendente (PKR), una proteina chinasi serina-treonina inducibile da interferone che porta all’interruzione della sintesi proteica come risposta all’infezione virale [89], la funzione principale degli RNA VAI adenovirali è la inibizione dell’attivazione di PKR [90].
Un recente rapporto ha suggerito che gli RNA VAI vengono trasformati in piccoli RNA e potrebbero comportarsi come siRNA funzionali o miRNA per regolare i componenti virali [91]. È interessante notare che l’oncogene Ras, che è sovraespresso in molti tumori, è anche in grado di inibire la PKR. Questa proprietà è stata utilizzata per costruire un adenovirus senza l’RNA virale associato, consentendo così la sua replicazione solo nelle cellule con sovraesprimono Ras [92].
Un altro rapporto indica che gli RNA VA sopprimono l’interferenza dell’RNA (RNAi) successivamente dopo l’infezione sopprimendo l’attività di due enzimi chiave, Dicer e il complesso di silenziamento indotto dall’RNA (RISC) [93].

Inibizione dell’Adenovirus VA RNA di entrambe le vie Interferone / PKR. I VA RNA (con struttura secondaria a stemloop imperfetto) inibiscono la segnalazione PKR indotta da IFN portando alla fosforilazione di eIF2α a valle e all’inibizione della traduzione proteica finale. La segnalazione di Ras tramite MEK e il suo substrato ERK può anche inibire l’attivazione di PKR indotta da IFN prevenendo la fosforilazione di PKR. Inoltre, i VA RNA possono anche essere trasformati in siRNA e inibire la funzionalità della via Dicer / RISC RNAi della cellula ospite, bloccando così la degradazione dei dsRNA virali corti e la via di sintesi endogena di miRNA / siRNA della cellula.
L’assemblaggio dell’adenovirus è attivato dall’incapsulamento del DNA virale che contiene un segnale di confezionamento ricco di AT all’estremità sinistra [94]. L’uscita delle particelle di adenovirus dal nucleo dove sono maturate è facilitata dalla rottura della membrana nucleare seguita dal collasso della membrana plasmatica senza necessariamente mostrare segni di apoptosi [81].
PROGETTARE COSTRUZIONI ADENOVIRALI UTILI PER LA TERAPIA GENICA DEL CANCRO
Diverse proprietà degli adenovirus li rendono utili come vettori per la terapia genica del cancro. Questi includono, la natura lieve della malattia derivante da infezioni da adenovirus, la mancanza di integrazione nel genoma dell’ospite, la loro elevata capacità di trasduzione.
Un adenovirus wild type ha una capacità “cargo” di 2 kb per un potenziale trasferimento genico, corrispondente a ~ 105% della dimensione originale del genoma [95].
Questa capacità è limitata dal volume intra-capside e dalle interazioni steriche con le proteine del nucleo virale al suo interno. Tuttavia, a seconda dell’applicazione, alcuni geni come E1 ed E3 possono essere eliminati per aumentare questa capacità a 7,5 kb [96]. Un ulteriore aumento della capacità a 11 kb può essere ottenuto con un’ulteriore cancellazione nella regione E2 [97].
Per gli adenovirus incompetenti di replicazione, chiamati anche adenovirus di prima generazione, le delezioni di geni adenovirali per produrre virus difettosi di replicazione sono anche uno sforzo per rendere il virus meno suscettibile alla generazione di particelle virioniche di tipo selvaggio.
Da un lato la delezione dei geni E1A porterà a una ridotta trascrizione dei geni E2, mentre la delezione di E1B migliorerà i segnali pro-apoptotici nelle cellule infette e infine la delezione di E3 ridurrà le possibilità di fuga di cellule infettate da virus risposte immunitarie [98, 99].
Queste delezioni limitano quindi l’uso di adenovirus nei protocolli di terapia genica a lungo termine, ma potenzialmente migliorano la loro applicazione nello sviluppo di vaccini. Gli adenovirus privi di geni E1 potrebbero essere coltivati in una linea cellulare di confezionamento trasformata con geni E1A ed E1B [100].
Questo sistema fornisce una preparazione virale superiore a 1013 particelle / ml, consentendo l’uso diretto in applicazioni in vivo [101]. Gli adenovirus con difetto di replicazione di seconda generazione sono stati successivamente sviluppati asportando alcuni o tutti i geni adenovirali dalle regioni E2 ed E4 [97, 102, 103].
Tuttavia, l’eliminazione di tutti questi componenti adenovirali non abroga la risposta immunitaria agli adenovirus, poiché la base di questa risposta non è solo innescata dal transgene terapeutico inserito nel genoma adenovirale [104] ma anche dai componenti strutturali richiesti dell’adenovirus [98, 105].
Le generazioni successive di vettori di adenovirus erano infatti mirate a rimuovere il numero massimo di geni dal genoma per generare il cosiddetto vettore “gutless” [106-108]. I vettori senza intestino contengono ITR e segnali di confezionamento ma richiedono un virus helper, il che significa che gli adenovirus senza intestino necessitano di cure speciali per la purificazione.
Questo problema è stato risolto disabilitando il confezionamento del virus helper attraverso l’uso del sistema cre-lox [109]. È emersa un’altra varietà di vettori, chiamati adenovirus ad alta capacità (HC-Ad) e in cui la maggior parte del DNA virale è stata sostituita da “stuffers” al fine di consentire un efficace confezionamento di adenovirus [106, 109-115].
Gli HC-Ad sono più adatti per la consegna genica terapeutica di geni di grandi dimensioni e, contrariamente ai loro predecessori di prima generazione, è stato segnalato che consentono l’espressione del transgene per un periodo di tempo sorprendentemente più lungo, oltre un anno [116].
ADENOVIRUS PER LA TERAPIA GENICA DEL CANCRO
L’uso di adenovirus per la terapia genica del cancro che coinvolge il rilascio di un gene terapeutico potrebbe essere classificato in tre categorie: 1) adenovirus che esprimono geni oncosoppressori; 2) adenovirus oncolitici potenzialmente armati di enzimi attivatori di profarmaci e; 3) adenovirus per vaccini a DNA. Tuttavia, è concepibile unificare tutte queste classi in una sola. Ad esempio, è possibile progettare un adenovirus oncolitico che sarà armato con un enzima che attiva il profarmaco e che potrebbe suscitare una risposta immunitaria. Un altro progetto potrebbe anche combinare geni oncosoppressori e geni immunomodulatori. Una vasta gamma di altre combinazioni è descritta in letteratura. Tuttavia, in questa recensione discuteremo solo le prime due categorie.
Adenovirus che esprimono geni oncosoppressori
Mutazioni o delezioni che portano alla perdita di funzione dei geni oncosoppressori e altri geni coinvolti nei checkpoint che controllano la qualità del materiale genetico, la divisione cellulare, la sopravvivenza e la morte delle cellule, spesso portano a uno squilibrio della crescita cellulare, che diventa non regolamentato, portando a un ciclo cellulare incontrollato. Tale squilibrio porta spesso allo sviluppo di cellule tumorali. Si ritiene che la reintroduzione di geni oncosoppressori o altri geni coinvolti nel controllo della crescita cellulare ripristini le normali funzioni cellulari [117].
Targeting del pathway p53: la chemioterapia anti-cancro che utilizza 60.000 composti contro un pannello di 60 linee cellulari tumorali umane ha dimostrato che è più efficace nelle linee cellulari tumorali che esprimono un gene p53 funzionale [118]. Tuttavia, la maggior parte dei tumori umani ospita un gene p53 difettoso [119]. Di conseguenza, è stato dimostrato che l’introduzione di un gene p53 wild-type nel carcinoma colorettale difettoso p53 porta alla soppressione della crescita cellulare [120]. Un recente studio clinico che utilizza il trasferimento del gene p53 mediato da adenovirus in pazienti con carcinoma esofageo avanzato resistente alle chemioterapia ha mostrato un chiaro effetto antitumorale [121]. Combinare il trasferimento di p53 con la chemioterapia è una buona pratica, che sinergizza le loro azioni. Per esempio, è stato scoperto che l’introduzione di p53 wild-type in cellule di cancro del polmone non di piccole dimensioni le sensibilizza al cisplatino [122] e sensibilizza le cellule di leucemia mieloide all’etoposide [123] e le linee cellulari di cancro della tiroide all’adriamicina [124]. La combinazione con la radioterapia è risultata ugualmente efficace sulle linee cellulari di cancro radioresistente del colon [125], dell’ovaio [126] e della testa e del collo radioresistenti in vivo [127]. Tuttavia, l’introduzione della funzione p53 wild type o la sua abrogazione non aumenta o riduce la radiosensibilità di alcune linee cellulari [128, 129], indicando una radiosensibilizzazione p53 specifica per tipo cellulare. La sovraespressione del doppio minuto 2 del topo (mdm2) in alcuni tumori potrebbe limitare l’efficacia del trasferimento del gene p53, ad esempio il 30% dei tumori dell’osteosarcoma sovraesprimono mdm2 a causa dell’amplificazione genica [130]. Questo è stato risolto attraverso la creazione di un p53 chimerico in cui sono stati sostituiti i domini che mediano la sua inattivazione [131]. Questo p53 chimerico ha indotto l’apoptosi sei volte in modo più efficiente rispetto a p53 wild type. Altre modifiche per superare l’inattivazione da parte di mdm2 sono le sostituzioni dei residui idrofobici Leu-14 e Phe-19 su p53, entrambi coinvolti nell’interazione con mdm2 [66]. La p53 modificata (14/19) è stata rilasciata da Ad-p53 (14/19) e si è scoperto che induce l’apoptosi in modo più drammatico rispetto alla p53 wild type, specialmente nelle cellule di osteosarcoma che sovraesprimono mdm2 [132]. Altre modifiche per superare l’inattivazione da parte di mdm2 sono le sostituzioni dei residui idrofobici Leu-14 e Phe-19 su p53, entrambi coinvolti nell’interazione con mdm2 [66]. La p53 modificata (14/19) è stata rilasciata da Ad-p53 (14/19) e si è scoperto che induce l’apoptosi in modo più drammatico rispetto alla p53 wild type, specialmente nelle cellule di osteosarcoma che sovraesprimono mdm2 [132]. Altre modifiche per superare l’inattivazione da parte di mdm2 sono le sostituzioni di residui idrofobici Leu-14 e Phe-19 su p53, entrambi coinvolti nell’interazione con mdm2 [66]. La p53 modificata (14/19) è stata rilasciata da Ad-p53 (14/19) e si è scoperto che induce l’apoptosi in modo più drammatico rispetto alla p53 wild type, specialmente nelle cellule di osteosarcoma che sovraesprimono mdm2 [132].
Il trasferimento di p53 wild type sarà probabilmente inefficace nel cancro cervicale indotto da HPV poiché la proteina E6 dell’HPV inattiva p53 attraverso la proteolisi mediata dall’ubiquitina [133], nelle cellule tumorali che esprimono l’antigene T grande SV40 in cui l’antigene T inattiva p53 [134] , o nel cancro del fegato indotto da epatite dove la proteina X esclude p53 dal nucleo [135].
Altri geni oncosoppressori: sono stati esplorati approcci che coinvolgono l’inibizione delle chinasi dipendenti dalla ciclina mediante rilascio adenovirale di p21 waf / cip1, un inibitore universale di CDK e un mediatore per l’arresto di p53 G1. Il trasferimento di p21 induce l’apoptosi [136] e sarebbe utile per quelle cellule tumorali che sovraesprimono mdm2 con conseguente p53 inattivato. Infatti è stato dimostrato che p21 era più efficace di p53 in un glioma di ratto [137]. In alcuni casi, tuttavia, la mancanza di p21 è alla base della maggiore sensibilità alla chemioterapia [138, 139], gettando così qualche dubbio sull’utilità del trasferimento del gene p21.
Le delezioni di p16INK4 si riscontrano in oltre il 50% dei gliomi [140]. Confrontando l’impatto del trasferimento di adenovirus di p53, p21 e p16, si è riscontrato che mentre p16 induceva un ritardo nella crescita del cancro alla prostata simile a p21, era più debole di p53 [141]. Tuttavia, sorprendentemente, l’introduzione di p16 in una linea cellulare di melanoma ha portato a una drammatica chemioresistenza a metotraxato, vinblastina e cisplatino [142]. Un altro soppressore tumorale che potrebbe essere considerato è il gene Retinoblastoma (Rb), che lega il fattore di trascrizione E2F quando ipofosforilato ma lo rilascia quando iperfosforilato, consentendo così l’ingresso del ciclo cellulare nella fase S [143]. Le delezioni del gene Rb sono presenti nel polmone, nella vescica, nella mammella, nell’osteosarcoma e in altri tumori. Dopo il trasferimento adenovirale del gene Rb a questi tumori,
p27Kip1 è un altro inibitore universale di CDK della stessa famiglia di p21 e media un arresto della crescita simile nella fase G1 [146]. p27 è stato confrontato con p21 in seguito al trasferimento mediato da adenovirus nel cancro al seno ed è stato riscontrato che diminuisce le attività CDK più di p21 [147] e inibisce la crescita tumorale del glioma in vivo [148]. Sembra tuttavia che l’approccio di fornire geni oncosoppressori utilizzando adenovirus con difetto di replicazione sia di utilità limitata poiché non tutte le cellule tumorali saranno infettate con successo.
Tra tutti i soppressori tumorali discussi qui, sembra che l’uso dei mutanti p53, resistenti all’inattivazione di mdm2 [131, 132] siano i candidati più probabili per procedere verso un uso clinico avanzato. Tuttavia, a causa della limitata diffusione imposta agli adenovirus con difetto di replicazione, sembra logico fornire p53 utilizzando adenovirus a replicazione condizionale (CRAD). Tuttavia, questo concetto presenta un potenziale problema. Poiché la maggior parte dei prodotti del gene E1 sono antagonisti di p53, specialmente E1B-55 kDa che interagisce direttamente con p53 e media la sua inattivazione [68] e traslocazione al citoplasma [69]. Anche in questo caso, E1B-55 kDa media anche l’ubiquitinazione e la degradazione di p53 in concerto con la proteina adenovirale E4orf6 [70, 71]. L’interazione di E1B-55 kDa e p53 infatti trasforma p53 in un potente repressore aumentando l’affinità di p53 al suo sito di legame [72]. Questa interazione suggerisce che E1B-55 kDa riduce l’attività soppressore del tumore di p53, che è l’obiettivo del trasferimento di p53 nelle cellule tumorali in primo luogo. Tuttavia, poiché l’E1B-55 kDa viene prodotto all’inizio del ciclo di replicazione degli adenovirus, sembra ragionevole porre il gene p53 sotto il controllo di un promotore tardivo. Questo obiettivo è stato raggiunto inserendo il cDNA di p53 nella cassetta di trascrizione delle fibre [149]. La produzione tardiva di p53 non ha compromesso il ciclo dell’adenovirus competente per la replicazione. Tuttavia, questa elegante ingegneria non ha mediato una forte funzione di soppressore del tumore p53 in questo contesto, nonostante il suo forte accumulo nucleare.
Le strategie di targeting del soppressore tumorale dovrebbero fornire un trattamento multimodale che coinvolga 1) la funzione del soppressore tumorale assicurando al contempo che tale proprietà sarà richiesta nelle cellule tumorali bersaglio; 2) l’attività oncolitica aggiuntiva del CRAD per consentire la diffusione del gene oncosoppressore alle cellule tumorali; e 3) la possibilità di combinare i primi approcci con la chemioterapia o la radioterapia.
Adenovirus a replica condizionale (CRAD)
Come molti virus a DNA, gli adenovirus hanno sviluppato meccanismi intricati per interrompere i principali effettori dei checkpoint cellulari, come p53, mdm2 e Rb, influenzando così diversi percorsi cellulari inclusa la progressione del ciclo cellulare in un modo che favorisce il completamento del ciclo adenovirale.
Gli adenovirus ottengono questo notevole exploit grazie a un repertorio molto limitato di geni i cui prodotti possono ottenere diverse interazioni proteina-proteina tramite più domini all’interno della stessa proteina adenovirale “Fig. (3,, 4) “. 4)”. Tuttavia le cellule normali piuttosto che le cellule tumorali sono il bersaglio evolutivo degli adenovirus. Pertanto, la biologia dell’adenovirus è stata prima dettata e modellata per funzionare nelle cellule normali.
Se i meccanismi utilizzati dagli adenovirus wild-type per sovvertire i processi delle cellule normali sono alterati dalla rimozione di geni specifici dal genoma dell’adenovirus, la replicazione dell’adenovirus modificato sarà consentita solo nelle cellule con processi di fatto sovvertiti. Sembra che nella maggior parte delle cellule tumorali quelle vie adenovirali mirate siano già difettose, dando così un segnale di luce verde alla replicazione degli adenovirus.
CRAD generati dalla delezione di geni adenovirali
Uno dei primi adenovirus oncolitici che è stato sviluppato per la terapia del cancro è stato Onyx-015 [84]. Questo adenovirus è stato costruito su una premessa semplice ma molto elegante. L’adenovirus wild type dopo aver infettato una cellula normale (diciamo una via aerea, cellula epiteliale), costringerà la cellula ad entrare nella fase S tramite uno dei suoi primi prodotti, la proteina E1A, che interagisce con la proteina Rb. Rb è solitamente strettamente associato ai fattori di trascrizione E2F e questa stretta associazione blocca la progressione del ciclo cellulare.
L’interazione di E1A e Rb rilascia fattori di trascrizione E2F sollevando così il blocco e consentendo alle cellule infette di progredire nella fase S. Suscitando tale progressione, viene attivato il meccanismo di replicazione della cellula infetta e la replicazione dell’adenovirus è un diretto beneficiario [152].
La risposta della cellula infetta è rapida e cerca di interrompere questo dirottamento innescando l’espressione di p53 (vedi prima) che dovrebbe portare all’apoptosi [153], eliminando così la cellula infetta e prevenendo la successiva diffusione virale. Questa risposta cellulare è, tuttavia, contrastata da un’altra proteina adenovirale E1B-55 kDa (vedi prima), che inattiva p53 [154]. Onyx-015 è privo di un E1B-55 kDa funzionale ed è quindi “teoricamente” incapace di completare il suo ciclo nelle cellule che possiedono un funzionale p53. Tuttavia, Onyx-015 sarà in grado di farlo se p53 non è funzionale, come nel caso della maggior parte dei tumori umani [119].
I rapporti iniziali hanno mostrato una maggiore sensibilità delle cellule prive di p53 [84]. La specificità basata su p53 di Onyx-015 è stata successivamente messa in discussione dalla scoperta che le cellule che possiedono p53 funzionale erano permissive a Onyx-015 [155]. Tuttavia, questo risultato potrebbe essere contrastato dalla possibilità di un deficit indiretto nel percorso p53 dovuto ad esempio alla perdita di p14ARF [156]; p14ARF regola verso il basso mdm2, una ubiquitina ligasi che degrada p53 (vedi prima), consentendo così alle cellule cancerose con p14ARF difettoso o mdm2 sovraespresso di comportarsi come cellule difettose di p53 anche con un gene p53 wild-type [157].
Inoltre, e probabilmente più dannoso per la teoria di Onyx-015, alcune linee cellulari tumorali richiedevano la proteina E1B-55 kDa per consentire la replicazione di Onyx-015 indipendentemente dal loro stato di p53 [158]. Dovremmo ricordare che oltre a inattivare il soppressore tumorale p53, la proteina E1B-55 kDa ha due funzioni tardive tra cui l’interruzione della sintesi della proteina ospite e il trasporto dell’mRNA tardivo adenovirale [159, 160].
Successivamente si è scoperto che il trasporto tardivo di mRNA è consentito in alcune linee cellulari tumorali ma non in altre e solo dove l’esportazione era consentita era consentita la replicazione di Onyx-015, definendo così il vero meccanismo alla base della selettività di 0nyx-015 [161]. O’Shea e colleghi hanno anche dimostrato che le cellule non permissive diventerebbero permissive dopo uno shock termico che consente il trasporto tardivo dell’mRNA, quindi la fenocopia delle funzioni E1B-55 kDa [162].
Il meccanismo antitumorale di Onyx-015 non è quindi universale. Altri adenovirus oncolitici sono stati generati eliminando sia E1B-55 kDa che E1B-19 kDa per i tumori nulli di p53 e Rb [78, 163], mediante delezione di VA-RNA per i tumori Ras positivi [92] o eliminando E1B-55 kDa e modificando il dominio di interazione E1A / Rb nella regione costante CR2 [164, 165] (Tabella 1).
Tabella 1
Adenovirus oncolitici. Alcuni virus acquisiscono selettività per la replicazione tumorale sulla base di nuove delezioni geniche all’interno del loro genoma virale; altri ottengono selettività in base all’induzione del promotore specifico associata al cancro.
| Adenovirus | Specificità del tumore | Test clinico | Rif |
|---|---|---|---|
| Tipo selvaggio | Nessuna | N / A | [14, 236] |
| dl1520 (ONYX-015) | E1B-55 kDa-delezione / inibizione di p53 | Fasi I – III | [84] |
| dl337 | Nessuna | N / A | [237] |
| dl118 | Delezione E1B / inibizione di p53 | N / A | [238] |
| hTERT-Ad | Induzione specifica del promotore hTERT | N / A | [239] |
| ONYX-411 | Promotore E2F-1 | N / A | [240] |
| AD.DF3-E1 | Promotore DF3 | N / A | [167] |
| CV787 | Probasina di ratto e promotori di PSA | Fase I | [241] |
| CV764 | Callicreina ghiandolare umana e promotori di PSA | N / A | [242] |
| CN706 | Promotore di PSA | Fase I | [19] |
DMINISTRAZIONE DI ADENOVIRUS ONCOLITICI PER LA TERAPIA DEL CANCRO
Onyx-015 è stato il primo adenovirus oncolitico a replica condizionale a essere testato in uno studio clinico. La sicurezza e l’efficacia antitumorale di questo tipo di adenovirus oncolitici è ora evidente. L’efficienza terapeutica degli adenovirus oncolitici dipenderà dalla via di somministrazione. La somministrazione intratumorale è di gran lunga la modalità più comune per la somministrazione di questi agenti.
Questa modalità aumenta il targeting e aumenta le possibilità di infezione delle cellule tumorali iniziali. L’iniezione intratumorale richiederebbe molto meno adenovirus rispetto a se iniettata per via endovenosa, intra-arteriosa o intraperitoneale. Queste ultime tre modalità richiedono un’enorme quantità di particelle adenovirali: ~ 2 × 1013 pfu.
Questo enorme carico potrebbe innescare una risposta immunitaria ed è spesso alla base dell’elevata frequenza di sintomi simil-influenzali osservati per i pazienti che ricevono gli adenovirus oncolitici attraverso queste vie.
Altri sintomi che accompagnano le iniezioni di adenovirus sono l’aumento delle transaminasi epatiche, l’iperbilirubnemia e l’aumento delle citochine infiammatorie (TNFα) e delle interleuchine IL-1 e 6 nonché dell’interferone γ [193, 196].
La tragica morte di Jesse Gelsinger 7 anni fa durante una sperimentazione clinica presso l’Università della Pennsylvania in cui ricevette 4 × 1013 pfu per il trattamento del deficit di ornitina decarbossilasi [216] mostra la necessità di generare adenovirus con ridotta immunogenicità e maggiore effetto oncolitico. L’iniezione intratumorale è risultata 1000 volte più efficace delle iniezioni sistemiche negli xenotrapianti tumorali [217].
Mentre l’iniezione intratumorale sarebbe l’ideale per i tumori solidi localizzati, per i tumori metastatici sarebbe necessaria un’iniezione sistemica, nel qual caso la presenza di anticorpi neutralizzanti preesistenti sarà un ostacolo al successo di questa strategia e una combinazione con un altro trattamento come la chemioterapia sarebbe consigliabile.
Questo problema è aggravato dall’assenza di un modello animale e quindi solo gli studi su pazienti umani in studi clinici aiuteranno a capire come risolvere questo problema.
link di riferimento: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3354698/
Ulteriori informazioni: S. Atasheva el al., “Terapia sistemica del cancro con adenovirus ingegnerizzato che elude l’immunità innata”, Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/… scitranslmed.abc6659



{kind=link}