









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



The central dogma of molecular biology states that DNA is transcribed into mRNA, which is subsequently translated into protein [8]. The flow of genetic information in time and space is orchestrated by complex regulatory mechanisms. Gene therapy represents the introduction of genetic material into an individual’s cells and biological tissues.
Techniques like insertion, alteration, or removal of genes are employed for correcting defective genes responsible for disease development, which then cures a disease or ameliorates the clinical status of a patient [9].
Several vectors have been utilized for gene therapy and they are generally classified as non-viral and viral vectors. Non-viral vectors possess several advantages compared to the viral vectors, including low host immunogenicity and potential for scale-up [10].
However, the success of non-viral gene therapy has been very limited in the past, primarily due to the barriers existing for plasmid DNA (pDNA) delivery, e.g., the necessity to cross the nuclear membrane before translation, the presence of antibiotic resistance genes in pDNA, and most importantly, the difficulty in controlling and regulating long-term expression.
The lack of control of long-term expression of pDNA poses a huge disadvantage in terms of the duration of treatment and possible side effects, which is in contrast to conventional drugs, where the treatment can be stopped instantaneously. These disadvantages of pDNA can possibly be overcome by using mRNA [11].
The mRNA carries genetic information from the DNA in the nucleus to the cytosol, where it is used by the ribosomes as a template for protein synthesis. As opposed to pDNA, the mRNA is efficacious in both mitotic and non-mitotic cells because mRNA exerts its function in the cytoplasm, hence its function is not dependent on active cell division.
Furthermore, unlike pDNA or viral vectors, mRNA does not contain additional foreign genes, which makes mRNA a safer vector. The challenge of long-term expression posed by pDNA can also be overcome by using mRNA, since mRNA mediates a rapid, transient expression of the encoded protein and the duration of the production is well-defined (usually a few days or weeks, depending on the specific mRNA platform).
This makes mRNA expression easier to control than the gene expression from pDNA and viral vectors [11]. In addition, the manufacturing of mRNA is cell-free, which strongly reduces the chance of mRNA contamination with bacterial components. This makes it easier to produce mRNA than pDNA under good manufacturing practice conditions [12].
Finally, vector-induced immunogenicity can be avoided for mRNA therapeutics, unlike for viral vectors or virus-like particles, which may elicit a specific immune response against the exposed viral proteins [13]. Specific therapeutic applications of mRNA, which are currently being explored include
- (i) vaccination against cancer and infectious diseases,
- (ii) protein-replacement therapy, and
- (iii) gene editing.
- Table 1 summarizes examples of ongoing clinical trials of mRNA-based therapeutic and prophylactic vaccine candidates.
Table 1
Examples of ongoing clinical trials of mRNA-based vaccines.
| mRNA | Mechanism of Action | Disease/Condition | Administration Route | Study Phase | Sponsor/Collaborator | National Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| Therapeutic mRNA | ||||||
| W_ova1 vaccine | Induction of an anti-tumor immune response | Ovarian cancer | Intravenous | Phase I | University Medical Center Groningen/BioNTech | NCT04163094 |
| CT7, MAGE-A3, and WT1 mRNA-electroporated Langerhans cells (LCs) | Electroporation of dendritic cells with antigen mRNA | Multiple Myeloma | Subcutaneous | Phase I | Memorial Sloan Kettering Cancer Center | NCT01995708 |
| Personalized Cellular mRNA | Immunization with DCs pulsed with mRNA encoded tumor antigens | Brain cancer/Neoplasm Metastases | Not specified | Phase I | Guangdong 999 Brain Hospital/Beijing, Tricision, Trinomab, Jinan University Guangzhou | NCT02808416 |
| Personalized mRNA | Immunization with DCs pulsed with personalized mRNA | Glioblastoma | Not specified | Phase I | Guangdong 999 Brain Hospital/Beijing Tricision, Trinomab, Jinan University Guangzhou | NCT02808364 |
| MiHA mRNA | Immunization with DCs loaded with MiHA mRNA | Hematological malignancies | Intravenous | Phase I Phase II | Radboud University/ZonMw: The Netherlands Organization for Health Research and Development Dutch Cancer Society | NCT02528682 |
| WT1-mRNA | Immunization with DCs electroporated with WT1-mRNA | Acute myeloid leukemia | Not Specified | Phase II | Zwi Berneman/Kom Op Tegen Kanker stichting tegen kanker Research Foundation – Flanders (FWO: Fonds Wetenschappelijk Onderzoek) | NCT01686334 |
| Human CMV pp65-LAMP mRNA | Immunization with DCs pulsed with CMV pp65-LAMP mRNA | Glioblastoma | Intradermal | Phase II | Gary Archer Ph.D./Duke University | NCT03927222 |
| Personalized mRNA | Personalized mRNA tumor vaccine encoding neoantigen | Advanced esophageal squamous carcinoma, gastric adenocarcinoma, pancreatic adenocarcinoma, colorectal adenocarcinoma | Subcutaneous | Enrolling | Changhai Hospital/Stemirna Therapeutics | NCT03468244 |
| mRNA[BI 1361849 (formerly CV9202)] | Not specified | Metastatic non-small cell lung cancer | Not specified | Phase I Phase II | Ludwig Institute for Cancer Research/Cancer Research Institute, New York City, Boehringer Ingelheim MedImmune, CureVac, PharmaJet | NCT03164772 |
| mRNA-5671/V941 | Not specified | Neoplasms, carcinoma, non-small-cell lung, pancreatic neoplasms, colorectal neoplasms | Intramuscular | Phase I | Merck Sharp & Dohme | NCT03948763 |
| mRNA-4157 | Immunostimulants | Solid tumors | Not specified | Phase I | Moderna/Merck Sharp & Dohme | NCT03313778 |
| mRNA-4157 | Immunotherapy with the personalized cancer vaccine | Cutaneous melanoma | Not specified | Phase II | Moderna/Merck Sharp & Dohme | NCT03897881 |
| Personalized mRNA | Encoding neoantigen | Esophageal cancer Non-small cell lung cancer | Subcutaneous | Enrolling | Stemirna Therapeutics/The First Affiliated Hospital of Zhengzhou University | NCT03908671 |
| mRNA-3704 | Alpha-galactosidase stimulants; Methylmalonyl CoA mutase stimulants; Protein synthesis stimulants | Methylmalonic acidemia, metabolism, inborn errors | Intravenous | Phase I Phase II | Moderna | NCT03810690 |
| mRNA-2416 | OX40 ligand modulators | Relapsed/Refractory solid tumor malignancies or lymphoma | Intratumoral | Phase I | Moderna | NCT03323398 |
| mRNA-2752 | IL36G protein stimulants; Interleukin 23 stimulants; OX40 ligand modulators | Relapsed/Refractory solid tumor malignancies or lymphoma | Intratumoral | Phase I | Moderna/AstraZeneca | NCT03739931 |
| Prophylactic mRNA | ||||||
| mRNA-1647, mRNA-1443 | Not specified | Cytomegalovirus | Not specified | Phase I | Moderna | NCT03382405 |
| mRNA-1893 | Not specified | Zika virus | Not specified | Phase I | Moderna/Biomedical Advanced Research and Development Authority | NCT04064905 |
| mRNA-1653 | A combined human metapneumovirus and human parainfluenza virus type 3 vaccine | Human metapneumovirus and Human Parainfluenzavirus | Not specified | Phase I | Moderna. | NCT03392389 NCT04144348 |
| mRNA-1944 | Encoding for an anti-Chikungunya virus monoclonal antibody | Chikungunya virus | Parenteral | Phase I | Moderna | NCT03829384 |
| mRNA-1653 | Immunostimulants | Metapneumovirus and Parainfluenza virus | Parenteral | Phase I | Moderna | NCT03392389 |
| CV7202 | Immunostimulants | Rabies | Intramuscular | Phase I | CureVac | NCT03713086 |
Two classes of mRNAs, i.e., non-replicating and self-amplifying mRNA, are commonly used as vaccine vectors. Non-replicating mRNA encodes only the protein antigen(s) of interest, while self-amplifying mRNA also encodes proteins enabling RNA replication [14]. Vaccines based on self-amplifying mRNA encode the RNA genome of a single-stranded RNA virus, e.g., an alphavirus, a flavivirus [15], or a picornavirus [7].
They are engineered to increase the duration and level of expression, as well as the subsequent immune response induced by the encoded antigen(s). They efficiently amplify the production of sub-genomic mRNA encoding antigen(s) of interest subsequent to a single round of replication. While both self-amplifying mRNA and non-replicating mRNA find application in prophylactic vaccines for infectious diseases, non-replicating mRNA is used for cancer vaccines
Fundamental Pharmacology of mRNA Vaccines
In vitro transcribed (IVT) mRNA is employed therapeutically as it mimics fully mature native mRNA present in the eukaryotic cytosol [16]. This may be achieved either by ex vivo transfection of cells with mRNA that are then adoptively transferred or by direct in vivo delivery of the IVT mRNA to the cytosol [17].
These approaches are explored for genome engineering, genetic reprogramming, adoptive T cell and dendritic cell (DC) based cancer and infectious disease immunotherapies, tolerization regimens to treat allergies, and protein replacement therapies.
Both ex vivo transfection and direct in vivo transfection enable the target cells to synthesize the encoded protein(s) in situ, where mRNA is used as a template and the protein(s) represents the active product. The open reading frame (ORF) of mature mRNA encoding the protein(s) of interest (the active product) marked by start and stop codons, respectively, is flanked by untranslated regions (UTRs), and ideally consists of a 5’ cap and a poly(A) tail [3].
The pharmacodynamic activity of both native and IVT mRNA takes place in the cytosol (Figure 1).
However, in contrast to endogenous mRNA, which is transcribed from DNA in the nucleus and enters the cytosol through nuclear export, IVT mRNA enters the cytosol from an extracellular source [18].
Once the IVT mRNA is delivered to the cytosol, its pharmacology is governed by the same complex cellular mechanisms that regulate the stability and translation of endogenous mRNA. The engineered IVT mRNA resembles the endogenous mRNA so closely that the cellular translation machinery is seamlessly utilized to synthesize a protein that may undergo post-translational modifications, eventually resulting in mature protein product(s).
In the case of vaccines, this mature protein product(s) represents the antigen(s), which may elicit potent pathogen-specific humoral and cell-mediated immune responses. However, the final intracellular destination is determined by the natural or engineered sequence(s) of the signal peptide or the transmembrane domain [7]. Therefore, mRNA vaccines can be designed for the delivery of the encoded protein(s) to the desired cellular compartment for proper presentation and/or function [19].

Mechanism of action of mRNA vaccines. 1. The mRNA is in vitro transcribed (IVT) from a DNA template in a cell-free system. 2. IVT mRNA is subsequently transfected into dendritic cells (DCs) via (3) endocytosis. 4. Entrapped mRNA undergoes endosomal escape and is released into the cytosol. 5. Using the translational machinery of host cells (ribosomes), the mRNA is translated into antigenic proteins. The translated antigenic protein undergoes post-translational modification and can act in the cell where it is generated. 6. Alternatively, the protein is secreted from the host cell. 7. Antigen protein is degraded by the proteasome in the cytoplasm. The generated antigenic peptide epitopes are transported into the endoplasmic reticulum and loaded onto major histocompatibility complex (MHC) class I molecules (MHC I). 8. The loaded MHC I-peptide epitope complexes are presented on the surface of cells, eventually leading to the induction of antigen-specific CD8+ T cell responses after T-cell receptor recognition and appropriate co-stimulation. 9. Exogenous proteins are taken up DCs. 10. They are degraded in endosomes and presented via the MHC II pathway. Moreover, to obtain cognate T-cell help in antigen-presenting cells, the protein should be routed through the MHC II pathway. 11. The generated antigenic peptide epitopes are subsequently loaded onto MHC II molecules. 12. The loaded MHC II-peptide epitope complexes are presented on the surface of cells, leading to the induction of the antigen-specific CD4+ T cell responses. Exogenous antigens can also be processed and loaded onto MHC class I molecules via a mechanism known as cross-presentation (not shown in the figure). The figure was created with BioRender.com.
The pharmacokinetics of IVT mRNA is determined by the half-life of the mRNA and the resulting mature protein after post-translational modification. The two major factors influencing the bioavailability of exogenous mRNA in the cytosol are
- (i) rapid RNase-mediated degradation and
- (ii) lack of passive diffusion across the plasma membrane owing to high molecular weight and electrostatic repulsion between the negative charges of the proteoglycan-coated cell membrane and the negatively-charged mRNA molecules [20].
Naked mRNA is rapidly degraded by extracellular RNases, thus hindering its efficient delivery and efficacy. A wide range of in vitro and in vivo transfection reagents have been shown to protect the mRNA against degradation and facilitate the cellular uptake and endosomal escape of mRNA.
Major efforts have been dedicated to improving the enzymatic RNA stability, as discussed further below [3]. Ultimately, the IVT mRNA composed of natural nucleotides is metabolized by inherent physiological mechanisms, hence reducing the risk of metabolite-induced toxicity.
Therefore, the delivery issues can be overcome by approaches including encapsulation of mRNA in drug delivery systems consisting of cationic molecules, lipids, polymers, and nanoparticles [21] as well as targeting DCs [22]. In addition, physical transfection methods like electroporation have been shown to enhance the delivery efficiency of large, self-amplifying mRNA in vivo, upon measuring reporter gene expression and immunogenicity of genes encoding HIV envelope proteins [23].
Approaches for Enhancing mRNA Stability
The development of mRNA-based drugs dates back to 1990 with the successful expression of a number of different proteins upon injecting mRNA encoding these proteins directly into the muscles of mice [24].
This led to
- (i) the testing of the first mRNA-based vaccine in 1993, which was shown to induce an anti-influenza cytotoxic T-lymphocyte response in mice [25], and
- (ii) the first vaccination with mRNA-encoding cancer antigens in 1995 [26].
These inceptive demonstrations ratified the potential of mRNA for
- (i) in situ expression of specific proteins and
- (ii) for induction of protective antigen-specific cellular and humoral immunity. However, the field was neglected for almost ten years until the potential of in vivo application of mRNA, i.e., induction of specific cytotoxic T lymphocytes and antibodies, was discovered [27].
Advances in the mRNA field were slow due to the labile nature of mRNA, which makes experiments employing unmodified mRNA very challenging unless precautions to handle mRNA are strictly adhered to [28]. Instead, the focus was directed towards DNA-based drugs, since DNA is more stable than RNA.
In a cell-free system, mRNA can be synthesized by IVT of a DNA template (e.g., a linearized plasmid or a PCR product), which encodes all the structural elements of a functional mRNA. To perform an IVT reaction, all the elements of the natural transcription process are required, i.e., a DNA template, an RNA polymerase, and nucleotide building blocks.
During the subsequent purification of the mRNA, the DNA template is often degraded by the addition of DNases, followed by purification by means of other conventional methods for isolating mRNA, e.g., precipitation and chromatography. This process results in highly pure mRNA products ready for use [29,30,31]. Several strategies have been pursued to cope with mRNA’s inherent lack of stability and potential immunogenicity, which are discussed further below.
Molecular Stabilization
Strategies including engineering of sequences and/or structure to enhance mRNA stability (extend the half-life) and translation are often instrumental in increasing the protein expression levels. Techniques employed to achieve this includes elongation of the poly(A) tail, modification of the 5’ cap, engineering of the UTRs and the sequence patterns in the ORF, and/or incorporation of modified nucleotides (Figure 2).

Structure of in vitro transcribed (IVT) mRNA and commonly used modification strategies. The design of IVT mRNA is based on the blueprint of eukaryotic mRNA, and it consists of a 5’ cap, 5’ and 3’ untranslated regions (UTRs), an open reading frame (ORF) encoding antigen(s), and a 3’ poly(A) tail. The IVT mRNA can be modified in one or multiple sites, e.g., by modification of the caps, the UTRs and/or the poly(A) tail, to modulate the duration and kinetic profile of protein expression. eIF4E, eukaryotic translation initiation factor 4E.
Cap Analog
A synthetic cap analogue can readily be added to the mRNA because the 5’ end cap is not encoded by the DNA template. Natural eukaryotic mRNA has a 7-methylguanosine (m7G) cap coupled to the mRNA during the transcription process via a 5’-5’-triphosphate bridge (ppp) [32].
The m7GpppN structure at the 5’ end of the mRNA cap serves several functions [33]. First, it protects the mRNA from rapid degradation by exonucleases. Second, it plays an indispensable role during translation because the eukaryotic initiation factor (eIF) 4E recognizes and binds to the cap of the mRNA.
It further plays a role in preventing innate immune sensors from recognizing the mRNA [34]. The mRNA may contain one of three distinct caps, i.e., cap-0 [m7G(5’)pppN1pN2p], cap-1 [m7G(5’)pppN1mpNp] and cap-2 [m7G(5’)pppN1mpN2mp], respectively [11].
Capping of IVT mRNA can be performed using two different approaches: The first approach includes the addition of a second step with recombinant vaccinia virus-derived capping enzymes after the transcription, resulting in a cap identical to the most frequent endogenous eukaryotic cap structure i.e., 7-methylguanosine (m7G) cap [30].
Alternatively, a synthetic cap analogue may be added during the in vitro transcription reaction, hence capping and in vitro transcription is performed in a single step. This approach is referred to as co-transcriptional capping [35]. A major drawback of this approach is the competition between the cap analog and the GTP nucleotide required for in vitro transcription, which ultimately results in a fraction of uncapped and translationally inactive mRNA [36]. The fraction of uncapped mRNA containing 5’ ppp groups is more immune-stimulating, which can be rectified by treatment with phosphatase to remove the ppp group at the 5’ end of uncapped mRNA [37].
Three different classes of m7GpppG cap analogs are used [38]:
- (i) anti-reverse cap analogs (ARCAs) [39],
- (ii) 3’-O-Me-m7GpppG [40], and
- (iii) modified ARCAs [41].
Initial mRNA research was performed using mRNA containing the m7 cap analog (GpppG) [42], and it is currently the most commonly used mRNA cap in clinical trials. Unfortunately, a fraction of the m7GpppG cap analog used during in vitro transcription becomes incorporated in the opposite orientation and is therefore not recognized by the ribosomes, eventually resulting in lower translational activity.
To avoid this, the so-called ARCA with only one 3’-OH group instead of two 3’-OH groups (ARCAs; m27,3’−OGpppG) has been introduced to prevent incorporation in the opposite orientation [43]. ARCAs juxtaposing the traditional cap analogues have been shown to exhibit more than four times RNA transcription efficiency [44].
In addition, the duration and the levels of protein expression have been found to be enhanced in cells transfected with ARCA-capped IVT RNA [41]. Recently, new types of chemically-modified cap analogues have been introduced, e.g., of phosphorothioate, phosphorothiolate [35], imidiphosphate [45], locked nucleic acid [46], boranophosphate bonds [47] and other types of modifications, which provide the mRNA with resistance to decapping by the mRNA-decapping enzyme 2, eventually resulting in a longer half-life of the mRNA [48].
Conventionally, the synthetic 5’-Cap 0-capped RNA is transcribed by performing an in vitro transcription, where more than 80% of the GTP added to the reaction is substituted with a dinucleotide cap analog (i.e., m7G[5’]ppp[5’]G), resulting in initiation of transcription with the cap analog [49].
However, this approach has been shown to exhibit a number of efficiency-related drawbacks, which have been overcome by the introduction of ScriptCap™. ScriptCap™ involves the addition of enzymatically built cap-0 structures onto the RNA transcripts by employing a capping enzyme with 100% reaction efficiency [50]. Native eukaryotic mRNA may contain a cap-1 or cap-2 but never a cap-0 [11].
Therefore, IVT mRNA should contain a cap-1 or a cap-2 in order to be less immune-stimulating [51]. A hallmark of these cap structures is the methylation status of the 2’ position of the 5’ second last and third last nucleoside. Prior to the advent of the novel technology of CleanCap™ introduced by TriLink, synthetic mRNA with a cap-1 could only be prepared by enzymatic capping [52,53].
However, cap-1 or cap-2 can now be incorporated during co-transcriptional capping at a capping efficiency of approximately 94% [52]. Notably, the capping efficiency has been shown to be significantly higher than the efficiency achieved by traditional co-transcriptional capping with cap-0 or ARCA [54].
5’ and 3’ Untranslated Regions (UTRs)
The importance of incorporation of 5’- and 3’-UTRs has been noted during in vitro post-transcriptional regulation of gene expression [55]. The numerous roles that UTRs play include
- (i) regulation of mRNA export from the nucleus,
- (ii) regulation of translation efficiency [56],
- (iii) orchestration of subcellular localization [57], and
- (iv) mRNA stability [58].
Introduction of α-globin 3’ end UTRs results in stabilization of mRNA, while the incorporation of beta-globin 5’ end and 3’ end UTRs leads to enhanced translational efficiency [59]. The optimal outcome is achieved by using two β-globin 3’-UTRs aligned in a head-to-tail configuration. α-globin and β-globin UTRs have been incorporated for tweaking the RNA for optimized in vitro transcription followed by mRNA electroporation of autologous T cells [60] and intranodal injection of naked antigen-encoding RNA [61].
Moreover, DCs transfected with antigen-encoding UTR-optimized mRNA have been used in a study involving immunization of cytomegalovirus-seropositive individuals and cancer patients [62]. In some situations, destabilizing the mRNA might be a viable approach to reduce the duration of protein synthesis. This may be accomplished by introducing adenylate-uridylate-rich elements in the 3’-UTRs of the mRNA, eventually resulting in faster mRNA degradation and shortening of the duration of protein expression [63].
Poly(A) Tail
The poly(A) tail plays a significant role in mRNA translation as well as for the enzymatic stability of mRNA. The poly(A) tail binds to several polyadenosyl binding proteins (PABPs) while working synergistically with 5’m7Gcap sequences to regulate translational efficiency [64].
Eukaryotic translation initiation factor eIF4E binds to the 5’m7G cap, which in turn complexes with eIF4G and eIF4A. PABP then interacts with the N-terminus of the eukaryotic translation initiation factor eIF4G, which forms an mRNP (messenger ribonucleoprotein) or a polysome complex [65].
The former depicts the mRNA-protein complex not yet involved in protein synthesis, while the latter is one that is already being translated. An adequately long poly(A) tail is required to circularize the mRNA via binding of PABPs to the poly(A) tail and the cap [55,66]. It has been observed that increasing the poly(A) tail length improves the efficiency of polysome generation and consequently influences the protein expression levels [67].
It has been shown that a gradual increase in the poly(A) tail length of IVT mRNA to 120 bases commensurately increases the protein expression level, while an increase in the number of bases beyond 120 does not further enhance protein expression [68]. Poly(A) tails can be added to mRNA by encoding the poly(A) tail in the DNA template, or by extension of the IVT RNA after transcription using recombinant poly(A) polymerase.
However, polyadenylation with recombinant poly(A) polymerase results in variable poly(A) tail length, thereby yielding polyadenylated mRNA with varying lengths. Therefore, the preferred approach is the generation of poly(A) tails with well-defined length from the mRNAs transcribed from poly(A) tail-encoding DNA templates [69].
The physical interactions between the 5’ and 3’ ends of mRNA take place between the cap and the poly(A) tail [70]. The poly(A) tail also plays a role in preventing decapping and mRNA degradation because removal or shortening of the poly(A) tail to less than 12 residues results in degradation of the mRNA through cleavage of the 5’ cap structure and 5’ to 3’ exonucleolytic digestion or 3’ to 5’ degradation [71].
Formulation Strategies
Despite the promising potential of mRNA-based vaccines, efficient intracellular delivery of mRNA to the cytosol continues to pose a major hurdle, especially for mRNA administered systemically.
The large molecular weight (105–106 Da) [21] and high negative charge density of mRNA impair the permeation of mRNA across cellular membranes. It is well known that the absorption of mRNA in the absence of a delivery system is extremely low, and the half-life of mRNA is approximately 7 h [72].
Moreover, mRNA is an inherently unstable molecule, which is highly prone to degradation by 5’ exonucleases, 3’ exonucleases, and endonucleases [73]. Consequently, delivery systems are imperative for intracellular delivery of mRNA to the therapeutic site of action in vitro as well as in vivo [8,74].
Different strategies have been investigated to improve RNA delivery, including improved injection strategies such as microinjections [75], RNA patches [76], gene gun-based administration [77], protamine condensation [78], RNA adjuvants [79], and encapsulation of RNA in nanoparticles consisting of lipids and/or polymers [80].
Generally, IVT mRNA for cytosolic delivery is formulated with a delivery system by mixing with a complexing agent [81], which can protect the mRNA against rapid degradation and facilitate cellular uptake.
Although the general dogma in the field is that efficient carriers are needed for substantially enhancing the in vivo transfection of mRNA, naked mRNA have been applied in many in vivo studies. Hence, the following section discusses the delivery of naked mRNA, followed by sections discussing vector-based mRNA delivery [82,83,84,85].
Naked RNA
The simplest administration strategy comprises intramuscular (i.m.) injection of naked mRNA, and proof of concept was originally demonstrated by in vivo reporter gene expression in mice [24]. Since then, the effectiveness of naked mRNA has been confirmed upon i.m. [86], subcutaneous (s.c.) [87] or intradermal (i.d.) [88] injections. I.d. and s.c. administration of mRNA has been shown to mediate healing of various skin diseases and to ameliorate wound healing by in situ expression of specific proteins in the skin [75,89].
Adopting this approach circumvents several obstacles otherwise associated with systemic administration of mRNA, e.g., clearance from the bloodstream via the liver, the kidneys, and the spleen [75]. Very efficient translation of the encoded protein has been shown for mRNA administered s.c. [90,91,92].
Interestingly, more efficient translation has at times been measured when compared to mRNA-loaded nanoparticle-based delivery systems [90,93]. Hence, it circumvents the need to employ carriers, eventually contributing to reduced cost and potential risk. Another advantage of administering mRNA-based vaccines via the s.c. route is that both cellular and humoral immune responses are induced [94] because the mRNA is expressed by both skin-resident DCs [95] and non-immune cells [96].
However, the outermost stratum corneum layer of the epidermis serves as a tight barrier to the absorption of topically administered drugs [97]. Heretofore, various approaches have been adopted to overcome this barrier, including physical (e.g., microporation [98], microneedles [99], and jet injection [100,101]), active (e.g., electroporation [102], iontophoresis [103], and sonophoresis [104]), and passive methods (e.g., nanoparticles [105] and liposomes [106]).
Electroporation and sonoporation can transiently permeabilize the skin by means of electric pulses and low-frequency ultrasound, respectively, for efficient delivery of genes into the skin [107]. Modified mRNA-encoding vascular endothelial growth factor-A was formulated in citrate-buffered saline without the use of a delivery system and was used for i.d. vaccination of patients with type 2 diabetes [108].
However, in spite of pronounced, sustained, dose-dependent and cargo-specific vasodilation, blood flow increase, oxygen-metabolic upregulation, angiogenesis and neovessel formation in animal models, the vasodilatory and angiogenic activity did not translate into humans [108,109].
Microneedle-based delivery is also an efficient technique where micron-sized needle patch/arrays composed of water-soluble polymeric or sugar excipients are employed, into which the mRNA is incorporated [76]. The patches/arrays provide mechanical strength needed for the needle to permeate the stratum corneum and penetrate into the viable skin layers.
Following injection, depending upon the type of microneedles, the patches/arrays degrade/dissolve and the encapsulated drug is released in the interstitial fluid of the skin [75]. Administration of mRNA by using dissolvable microneedles provides an important advantage, i.e., delivery in a solid dosage form circumvents the necessity of dealing with mRNA in a liquid dosage form [110], which therefore eliminates the menace emanating from RNase contamination, along with increasing mRNA stability, and shelf life [76].
Scavenger receptor-mediated endocytosis and micropinocytosis have been shown to be the active uptake mechanisms for naked mRNA in immature DCs [16]. However, naked mRNA displays a short plasma half-life, is prone to ribonuclease degradation, and faces difficulties in entering the cell. Therefore, delivery systems have been propounded to protect the mRNA and shield its negative charge.
Viral Vectors
Delivery of mRNA can be mediated by viral and non-viral vectors. Non-viral vectors can further be categorized into lipid-based delivery systems, polymer-based delivery systems, and lipid-polymer hybrid systems [111]. For viral RNA delivery, there has been a great deal of interest in the engineering of adeno-associated viruses to carry nucleic acid cargoes [112].
Genetically-modified viruses are usually employed for mRNA/gene delivery. The genes of these viruses are partially or fully substituted with model or therapeutic genes. A benefit of RNA viruses is that they are replicated and expressed locally in the cytoplasm.
Positive strand RNA viruses are distinguished by a genomic sequence that can be translated directly into proteins of interest by host ribosomes. Notably, alphaviruses (e.g., Sindbis and Semliki Forest virus) [113], picornaviruses [114], and flavivirus [115] (e.g., Kunjin virus) have been employed for mRNA delivery. Various alphavirus vectors can be used to express high levels of exogenous protein in a wide spectrum of hosts [116].
Commonly used approaches include the direct substitution of structural genes with heterologous expression or placing the nonstructural genes downstream of the RNA sub-genomic promoter [117]. However, alphaviruses induce severe cytopathogenic effects, which restrict their application in gene therapy, although different strategies can be employed to surmount this challenge [118].
Some of these strategies include engineering of mutant vectors with mitigated cytotoxicity and temperature-inducibility, and self-inactivating vectors with point mutations in the nsP2 gene (especially at position 726, 259 and 650) [119]. Sendai virus (SeV) (murine parainfluenza virus type 1 or hemagglutinating virus of Japan) that belongs to the Paramyxoviridae family is worth mentioning owing to its popular application as a vector.
It is favored for its high but transient gene expression levels, wide host cell specificity, low pathogenicity, and strong immunogenicity [120]. As a vaccine platform, the Venezuelan encephalitis virus is of particular interest [121]. These vaccines encompass the live-attenuated viral vaccine TC-83 and a formalin-inactivated variety of it is referred to as C-84, which boosts the efficacy and increases the duration of immunity upon administering the TC-83 vaccine via different administration routes, intranasal being the most widely used [122].
However, using viral vectors embodies crucial drawbacks associated with genome integration, and possible host rejection (immunogenicity and cytotoxicity) among others [123], hence provoking the need for non-viral vectors for mRNA delivery [10].
Polymer-Based Vectors
Diethylaminoethyl (DEAE) dextran was the first polymer to be tested as a delivery reagent for IVT mRNA [124]. Later, it was shown that lipid-mediated mRNA transfection is 100 to 1000 times more efficient than DEAE-dextran [125]. This discovery stalled the progress of polymeric carriers and paved the way for lipid-based transfection reagents for nucleic acids, including mRNA.
A comprehensive study compared the polymers poly-beta-amino-esters (PBAE) and polyethylenimine (PEI) with commercial transfection reagent Lipofectamine™ 2000 and 1,2-dioleoyl-3-trimethylammonium propane (DOTAP)/ 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) for functional, antigen-specific T-cell responses after mRNA delivery [126].
All carriers were complexed with mRNA encoding the HIV-1 antigen gag. Gag-specific, IFN-γ secreting T cells were measured in the spleen and lymph nodes of mice immunized with gag mRNA complexed with cationic lipids but not in mice immunized with naked and polymer-complexed mRNA. PEI and its derivatives are among the most commonly employed cationic polymers [127].
They are water-soluble, display a high density of positive charge associated with the amino groups, and are proven mRNA carriers for in vitro transfection [128]. However, PEI displays toxicity issues owing to the high molecular weight (>25 kDa), which may arise from the adsorption of anionic serum proteins onto the polyplex surface between cationic polymers and anionic serum plasma proteins.
However, the resultant increase in size is only transient as the proteins adsorbed on the surface of polyplexes prevent particle–particle aggregation in the long run [129]. Various efforts have been made to mitigate these challenges. The first proof of concept for safe and efficacious mRNA vaccine transfection employing cationic polymer was obtained by intranasal administration of 2 kDa PEI conjugated to cyclodextrin.
Cyclodextrin conjugated to PEI enabled delocalization of the charge density on the polyamine backbone, hence reducing cytotoxicity and at the same time maintaining protonatable groups, resulting in improved transfection [130]. Polymeric nanoparticles composed of biodegradable polymers, e.g., poly(lactic-co-glycolic acid) (PLGA), are well suited for incorporation of hydrophobic and positively-charged molecules. They provide good colloidal stability, low toxicity, and the possibility of sustained release. However, due to the anionic nature of PLGA at physiological pH [131], the mRNA encapsulation efficiency is very low.
Polymer-based carriers exhibit considerable potential for gene therapy owing to the substantial transfection efficiency and tolerable toxicity [132]. A series of multifunctional block copolymers, i.e., dimethylaminoethyl methacrylate (DEAMA), poly(ethylene glycol) methacrylate, and DEAEMA-co-n-butyl methacrylate, demonstrated a transfection efficiency of 77% and 50% in RAW 264.7 macrophages and DC2.4 dendritic cells, respectively, thereby exhibiting potential as a carrier for mRNA-based intracellular vaccine delivery [133].
While different types of polymers and copolymers have been tested, the correlation between the structure of polymers and their biological response, e.g., transfection and toxicity, was found to be poor and thus, design of various polymer-based delivery systems relies on empirical, rather than rational approaches [134]. Despite the advantages mentioned above, polymer-based delivery systems are not as clinically advanced as lipid-based delivery systems owing to their polydispersity and challenges pertaining to metabolism of large molecular weight polymers [21].
Lipid-Based Vectors
Vectors based on lipids or lipid-like compounds (lipidoids) represent the most commonly used non-viral gene carriers [21]. Various synthetic and naturally-derived lipids have been employed to form liposomes (complexes of liposomes and nucleic acids are referred to as lipoplexes) or lipid nanoparticles (LNPs), both of which have been reported to efficiently deliver mRNA-based vaccines (Table 2).
LNPs are often formulated by using cationic lipids displaying tertiary or quaternary amines to encapsulate the polyanionic mRNA. Cationic lipids spontaneously encapsulate negatively-charged mRNA, mediated by a combination of attractive electrostatic interactions with RNA and hydrophobic interactions, and thus have been used alone or in combination for lipofection of mRNA.
The first reported use of LNPs as delivery system for mRNA came in 2015, with the delivery system consisting of ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG-lipid in the ratio of (50:10:38.5:1.5 mol/mol) [86]. Examples of cationic lipids include e.g., 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) [135], DOTAP [136], and zwitterionic DOPE [137,138].
They are structurally denoted by a cationic headgroup, a hydrophobic tail group, and a linking group in between [139].
However, cationic lipids have been observed to exhibit pro-inflammatory reactions and undesirable side effects [140].
Therefore, neutral lipids are also incorporated into cationic liposomes to decrease toxicity and attain high transfection levels in vivo [106].
The mechanism of LNP-mediated delivery of mRNA is not fully understood, but LNPs are suggested to be internalized by endocytosis and are attached electrostatically and fused with the cell membrane via inverted non-bilayer lipid phases [21].
Table 2
Examples of nanoparticulate drug delivery systems for mRNA delivery.
| Drug Delivery Systems | Composition | RNA | Disease/Condition | References |
|---|---|---|---|---|
| Polymers | Poly(glycoamidoamine) | Erythropoietin (EPO) mRNA | Anemia and myelodysplasia | [168] |
| Polyethyleneimine | HIV-1 gag mRNA | HIV | [169] | |
| Poly(β-amino ester) (PBAE) | eGFP mRNA | N/A | [170] | |
| Triblock copolymer (comprising DMAEMA, PEGMA, DEAEMA and BMA) | eGFP and ovalbumin | N/A | [171] | |
| DEAE-Dextran | Luciferase-encoding mRNA | N/A | [172] | |
| Lipids | DOTAP/DOPE | HxB-2 HIV-1 Gag antigen mRNA | HIV | [126] |
| DOPE/DC-Cholesterol [2:1] | eGFP mRNA | N/A | [106] | |
| DOTAP/Cholesterol [1:1] liposome with DSPE-PEG and DSPE-PEG-AA | HSV I Thymidine kinase mRNA | Cancer | [142] | |
| C12-200:Cholesterol: DOPE:C14-PEG2000 | EPO mRNA | N/A | [157] | |
| A18 | Ovalbumin mRNA | Melanoma | [173] | |
| cKK-E12 | HER2 antibody mRNA | Cancer | [174] | |
| (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino)butanoate (MC3), DSPC, cholesterol, and 1,2-dimyristoyl-rac-glycerol, methoxypolyethylene glycol (PEG2000-DMG) | Human erythropoietin | [175] | ||
| DOTAP/DOPE [1:1] | HIV-1 antigen Gag mRNA | HIV | [126] | |
| 3β-[N-(N’,N’-dimethylaminoethane) carbamoyl](DC-Cholesterol)/DOPE (1:2) | eGFP mRNA | N/A | [106] | |
| Lipid polymer hybrid NPs | TT3:DOPE:Cholesterol:DMG-PEG2000 with PLGA core | Firefly luciferase (FLuc) mRNA and eGFP mRNA | N/A | [160] |
| PBAE:C14-PEG2000 | FLuc mRNA | N/A | [161] | |
| PBAE:EDOPC/DOPE/DSPE-PEG | Ovalbumin mRNA | N/A | [176] | |
| PBAE: DOPC, DOTAP, and DSPE-PEG | eGFP mRNA | N/A | [105] | |
| Peptides and peptide-polymer hybrids | PepFect14 | eGFP mRNA | Ovarian cancer | [164] |
| RALA | eGFP mRNA OVA mRNA | N/A | [165] | |
| RALA-PLA | eGFPmRNA FLuc mRNA | N/A | [167] |
Liposomes are closed membrane structures, which are formed by self-assembly when phospholipids are dispersed in aqueous systems [141]. They consist of at least one phospholipid bilayer, which mimics the cell membrane structure enclosing an aqueous core [8]. DOTAP/DOPE at a 1:1 molar ratio has been reported as an effective transfection agent for mRNA encoding the HIV-1 antigen Gag, which successfully induced an antigen-specific immune response in vivo in mice [126].
Additionally, 3β-N-(N’,N’-dimethylaminoethane) carbamoyl-Cholesterol)/DOPE-based liposomes in a [1:2] ratio achieved high encapsulation efficiency of enhanced green fluorescent protein (eGFP) mRNA, along with high eGFP expression in vitro [106]. Furthermore, an additional multi-component LNP displayed a tumor-suppressant effect when loaded with herpes simplex virus I (HSV I) thymidine kinase encoding mRNA.
The LNPs were composed of DOTAP/Cholesterol [1:1] liposomes along with 1,2-distearoyl-phosphatidylethanolamine (DSPE)-polyethylene glycol (PEG) and DSPE-PEG-anisamide (AA) [142]. The principle behind their effectiveness may be summarized as a combination of its electrostatic interactions attributable to opposite charges and hydrophobic interactions with mRNA.
Additionally, the endosomal escape capabilities and self-assembling properties resulting in uniform layers enclosing polymeric cores also contribute to the wide application of cationic lipids [143]. However, in vivo studies are more challenging due to the fast elimination of cationic lipids by the mononuclear phagocytic system [144].
Cationic lipids consisting of only one quaternary ammonium headgroup pose safety issues such as toxicity and immunogenicity in vitro [145] and in vivo [146]. For instance, cationic liposomes when administered via the intravenous route may induce hepatotoxicity [147] and can trigger a strong IFN-γ response in mice resulting in inflammation [148,149].
Furthermore, positively-charged lipids, e.g., DOTAP and DOTMA, can be neutralized by anionic serum proteins, leading to toxicity and reduced efficacy [150]. Moreover, challenges like unrestricted protein binding, colloidal instability, and drug leakage may arise [151].
Alternatively, new gene delivery vectors containing ionizable lipids [152] and lipid-like materials termed lipidoids [153] have been introduced to overcome challenges posed by conventional cationic lipids while retaining their advantageous transfection properties.
Ionizable lipids for mRNA transfection are positively-charged at low pH (which aids in mRNA complexation when it is carried out in acidic buffer) but are neutral at physiological pH (for reduced toxicity post-injection) [154]. Unlike conventional cationic lipids, these lipidoids display a series of secondary and tertiary amines allowing for more efficient interactions with mRNA without remarkably increasing the overall charge of the delivery system [155].
Encapsulation of mRNA in nanoparticles serves to physically protect nucleic acids from degradation and depending on the specific chemistry can aid in cellular uptake and endosomal escape [156]. The combination of the ionizable lipid C12-200, cholesterol, DOPE and C14-PEG2000 at a 3.5:4.65:1.6:0.25 molar ratio, respectively, encapsulating erythropoietin mRNA (EPO-mRNA) displayed high efficacy in vivo when injected into mice, measured as the cellular expression of EPO [157]. The emphasis on the nanoparticle platform for mRNA delivery is in part due to the application of established DNA and siRNA delivery systems.
Lipid-Polymer Hybrid Nanoparticles
Lipid-polymer hybrid nanoparticles (LPNs) have been demonstrated previously to exhibit effective functional delivery of siRNA in vitro [158] and therapeutic delivery of siRNA in vitro and in vivo [159]. This hybrid delivery system has also shown promising results for delivery of mRNA, with the mRNA being encapsulated in a hybrid nanoparticle composed of the lipid-like material N1, N3, N5-tris(2-aminoethyl)benzene-1,3,5-tricarboxamide (TT) in TT3:DOPE:Cholesterol:DMG-PEG2000 (1,2-dimyristoyl-sn-glycerol, methoxypolyethylene glycol) with a polymeric PLGA core [160]. In addition, optimized LPNs consisting of the degradable polymer PBAE, formulated with PEG-Lipid C14-2000, showed successful delivery of mRNA to the lungs [161].
This, along with reported co-delivery of siRNA and mRNA with lipidoid polymer hybrid nanoparticles [162], shows that LPNs are an emerging nucleic acid delivery system. The hybrid formulation is thermodynamically favorable, with respect to hydrophobic, van der Waal, and electrostatic interactions [80].
Several lipids and polymers have been investigated to formulate stable nucleic acid lipid particles using this delivery system. Most common polymers employed are PLGA, polycaprolactone, polylactic acid, or their combinations, whereas the lipids used include DOTAP, 1,2-dilauroyl-sn-glycero-3-phosphocholine, 1,2-distearoyl-sn-glycero-3-phosphocholine, lecithin, DSPE, and PEG, among others. Structurally, based on small-angle X-ray scattering and cryogenic transmission electron microscopy, these nanoparticles are suggested to entail a polymeric matrix core with lamellar lipid structures with the nucleic acid localized in the core and in the corona [163].
Peptide-Based Vectors
Peptide-based systems for mRNA delivery are gaining momentum due to the versatility peptides can offer. Peptide-based delivery systems, both alone and in combination with other materials such as polymers, have been reported in the literature. In a study concerning ovarian cancer therapy, the commercially available cell-penetrating peptide (CPP) PepFect14 was complexed with eGFP mRNA via attractive electrostatic interactions [164].
This nanoparticulate formulation was more efficient in transfecting eGFP mRNA into cells associated with ovarian cancer than commercially available lipofectamine MessengerMAX. Similarly, the CPP RALA has been used to effectively deliver both eGFP and OVA mRNA and has been demonstrated to outperform the cationic lipid DOTAP and the fusogenic lipid DOPE [165]. However, current limitations include targeted cell delivery [164] and short circulation half-life due to the low stability in serum-containing medium [166].
Recently, a novel polymer-peptide hybrid mRNA delivery nanoplatform was introduced [167] combining both polymer (PLA) based micelles and a cationic fusogenic peptide (RALA) to achieve appropriate degradability, mRNA stability, and endosomolytic properties for translation. It was reported to protect eGFP as well as FLuc mRNA against serum nuclease degradation and achieve DC transfection. Indeed, peptide-based vectors and hybrids are promising and interesting additions to the various existing non-viral carriers for the delivery of mRNA.
Cell-Specific mRNA Delivery
Cell-specific delivery of mRNA would be beneficial for the development of mRNA-based therapeutics. This can enhance the delivery of mRNA molecules to the targeted cells and hence reduce the required mRNA dose, as well as reducing potential off-target effects. It has been reported that lymphoid organs can be targeted by adjusting the net charge of the formulation [177]. This is based on the principle of APCs being in the vicinity of T cells in these organs, thus providing optimal conditions for efficient priming and amplification of T-cell responses.
Site-specific delivery of mRNA-loaded nanoparticles via active targeting has been shown to result in induction of strong effector and memory T-cell responses, and mediation of potent IFN-α-dependent rejection of progressive tumors, as observed with RNAs. In another study, cell-specific delivery of FLuc and IL-10 mRNA to leukocytes (Ly6c+) was achieved by coating the formulated mRNA-containing LNPs with anti-L6c+ monoclonal antibodies [178].
Alternatively, DCs and macrophages express receptors with the ability to present antigens, e.g., C-type lectin receptors [179], which recognize sugar groups such as mannose- and fucose-terminated glycans [180] and mediate the endocytosis of mannose-modified nanoparticles. This has been exploited for transfection of GFP mRNA into DCs by self-assembly of mannose-cholesterol conjugates with varying PEG units as linkers [181].
reference link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7076378/
Optimization of mRNA translation and stability
This topic has been extensively discussed in previous reviews14,15; thus, we briefly summarize the key findings (Box 1). The 5′ and 3′ UTR elements flanking the coding sequence profoundly influence the stability and translation of mRNA, both of which are critical concerns for vaccines.
These regulatory sequences can be derived from viral or eukaryotic genes and greatly increase the half-life and expression of therapeutic mRNAs23,24. A 5′ cap structure is required for efficient protein production from mRNA25. Various versions of 5′ caps can be added during or after the transcription reaction using a vaccinia virus capping enzyme26 or by incorporating synthetic cap or anti-reverse cap analogues27,28.
The poly(A) tail also plays an important regulatory role in mRNA translation and stability25; thus, an optimal length of poly(A)24 must be added to mRNA either directly from the encoding DNA template or by using poly(A) polymerase. The codon usage additionally has an impact on protein translation.
Replacing rare codons with frequently used synonymous codons that have abundant cognate tRNA in the cytosol is a common practice to increase protein production from mRNA29, although the accuracy of this model has been questioned30. Enrichment of G:C content constitutes another form of sequence optimization that has been shown to increase steady-state mRNA levels in vitro31 and protein expression in vivo12.
Although protein expression may be positively modulated by altering the codon composition or by introducing modified nucleosides (discussed below), it is also possible that these forms of sequence engineering could affect mRNA secondary structure32, the kinetics and accuracy of translation and simultaneous protein folding33,34, and the expression of cryptic T cell epitopes present in alternative reading frames30. All these factors could potentially influence the magnitude or specificity of the immune response.
*-*-*-*-*-*-*-
Box 1: Strategies for optimizing mRNA pharmacology
A number of technologies are currently used to improve the pharmacological aspects of mRNA. The various mRNA modifications used and their impact are summarized below.
- • Synthetic cap analogues and capping enzymes26,27 stabilize mRNA and increase protein translation via binding to eukaryotic translation initiation factor 4E (EIF4E)
- • Regulatory elements in the 5′-untranslated region (UTR) and the 3′-UTR23 stabilize mRNA and increase protein translation
- • Poly(A) tail25 stabilizes mRNA and increases protein translation
- • Modified nucleosides9,48 decrease innate immune activation and increase translation
- • Separation and/or purification techniques: RNase III treatment (N.P. and D.W., unpublished observations) and fast protein liquid chromatography (FPLC) purification13 decrease immune activation and increase translation
- • Sequence and/or codon optimization29 increase translation
- • Modulation of target cells: co-delivery of translation initiation factors and other methods alters translation and immunogenicity
*-*-*-*-*-*-*-
Modulation of immunogenicity
Exogenous mRNA is inherently immunostimulatory, as it is recognized by a variety of cell surface, endosomal and cytosolic innate immune receptors (Fig. 1) (reviewed in Ref. 35). Depending on the therapeutic application, this feature of mRNA could be beneficial or detrimental. It is potentially advantageous for vaccination because in some cases it may provide adjuvant activity to drive dendritic cell (DC) maturation and thus elicit robust T and B cell immune responses. However, innate immune sensing of mRNA has also been associated with the inhibition of antigen expression and may negatively affect the immune response9,13. Although the paradoxical effects of innate immune sensing on different formats of mRNA vaccines are incompletely understood, some progress has been made in recent years in elucidating these phenomena.
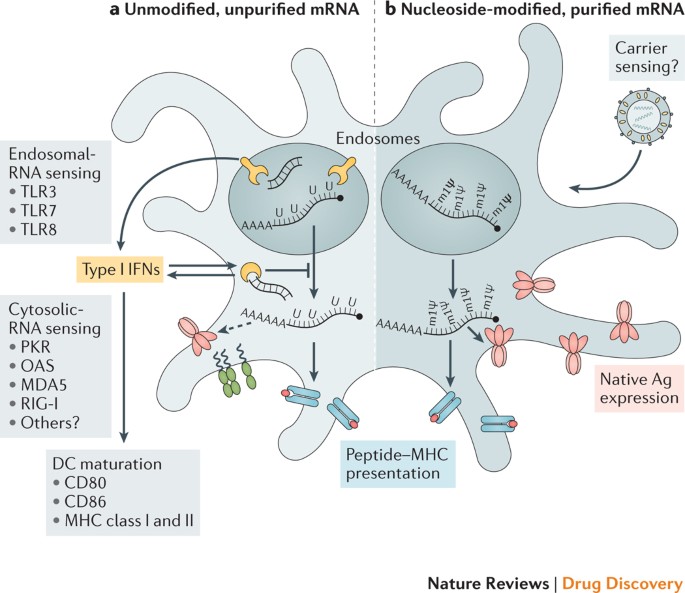
Studies over the past decade have shown that the immunostimulatory profile of mRNA can be shaped by the purification of IVT mRNA and the introduction of modified nucleosides as well as by complexing the mRNA with various carrier molecules9,13,36,37.
Enzymatically synthesized mRNA preparations contain double-stranded RNA (dsRNA) contaminants as aberrant products of the IVT reaction13. As a mimic of viral genomes and replication intermediates, dsRNA is a potent pathogen-associated molecular pattern (PAMP) that is sensed by pattern recognition receptors in multiple cellular compartments (Fig. 1).
Recognition of IVT mRNA contaminated with dsRNA results in robust type I interferon production13, which upregulates the expression and activation of protein kinase R (PKR; also known as EIF2AK2) and 2′-5′-oligoadenylate synthetase (OAS), leading to the inhibition of translation38 and the degradation of cellular mRNA and ribosomal RNA39, respectively. Karikó and colleagues13 have demonstrated that contaminating dsRNA can be efficiently removed from IVT mRNA by chromatographic methods such as reverse-phase fast protein liquid chromatography (FPLC) or high-performance liquid chromatography (HPLC). Strikingly, purification by FPLC has been shown to increase protein production from IVT mRNA by up to 1,000-fold in primary human DCs13.
Thus, appropriate purification of IVT mRNA seems to be critical for maximizing protein (immunogen) production in DCs and for avoiding unwanted innate immune activation.
Besides dsRNA contaminants, single-stranded mRNA molecules are themselves a PAMP when delivered to cells exogenously. Single-stranded oligoribonucleotides and their degradative products are detected by the endosomal sensors Toll-like receptor 7 (TLR7) and TLR8 (Refs 40,41), resulting in type I interferon production42.
Crucially, it was discovered that the incorporation of naturally occurring chemically modified nucleosides, including but not limited to pseudouridine9,43,44 and 1-methylpseudouridine45, prevents activation of TLR7, TLR8 and other innate immune sensors46,47, thus reducing type I interferon signalling48. Nucleoside modification also partially suppresses the recognition of dsRNA species46,47,48.
As a result, Karikó and others have shown that nucleoside-modified mRNA is translated more efficiently than unmodified mRNA in vitro9, particularly in primary DCs, and in vivo in mice45. Notably, the highest level of protein production in DCs was observed when mRNA was both FPLC-purified and nucleoside-modified13. These advances in understanding the sources of innate immune sensing and how to avoid their adverse effects have substantially contributed to the current interest in mRNA-based vaccines and protein replacement therapies.
In contrast to the findings described above, a study by Thess and colleagues found that sequence-optimized, HPLC-purified, unmodified mRNA produced higher levels of protein in HeLa cells and in mice than its nucleoside-modified counterpart12. Additionally, Kauffman and co-workers demonstrated that unmodified, non-HPLC-purified mRNA yielded more robust protein production in HeLa cells than nucleoside-modified mRNA, and resulted in similar levels of protein production in mice49. Although not fully clear, the discrepancies between the findings of Karikó9,13 and these authors12,49 may have arisen from variations in RNA sequence optimization, the stringency of mRNA purification to remove dsRNA contaminants and the level of innate immune sensing in the targeted cell types.
The immunostimulatory properties of mRNA can conversely be increased by the inclusion of an adjuvant to increase the potency of some mRNA vaccine formats. These include traditional adjuvants as well as novel approaches that take advantage of the intrinsic immunogenicity of mRNA or its ability to encode immune-modulatory proteins.
Self-replicating RNA vaccines have displayed increased immunogenicity and effectiveness after formulating the RNA in a cationic nanoemulsion based on the licensed MF59 (Novartis) adjuvant50. Another effective adjuvant strategy is TriMix, a combination of mRNAs encoding three immune activator proteins: CD70, CD40 ligand (CD40L) and constitutively active TLR4.
TriMix mRNA augmented the immunogenicity of naked, unmodified, unpurified mRNA in multiple cancer vaccine studies and was particularly associated with increased DC maturation and cytotoxic T lymphocyte (CTL) responses (reviewed in Ref. 51). The type of mRNA carrier and the size of the mRNA–carrier complex have also been shown to modulate the cytokine profile induced by mRNA delivery.
For example, the RNActive (CureVac AG) vaccine platform52,53 depends on its carrier to provide adjuvant activity. In this case, the antigen is expressed from a naked, unmodified, sequence-optimized mRNA, while the adjuvant activity is provided by co-delivered RNA complexed with protamine (a polycationic peptide), which acts via TLR7 signalling52,54.
This vaccine format has elicited favourable immune responses in multiple preclinical animal studies for vaccination against cancer and infectious diseases18,36,55,56. A recent study provided mechanistic information on the adjuvanticity of RNActive vaccines in mice in vivo and human cells in vitro54.
Potent activation of TLR7 (mouse and human) and TLR8 (human) and production of type I interferon, pro-inflammatory cytokines and chemokines after intradermal immunization was shown54. A similar adjuvant activity was also demonstrated in the context of non-mRNA-based vaccines using RNAdjuvant (CureVac AG), an unmodified, single-stranded RNA stabilized by a cationic carrier peptide57.
Progress in mRNA vaccine delivery
Efficient in vivo mRNA delivery is critical to achieving therapeutic relevance. Exogenous mRNA must penetrate the barrier of the lipid membrane in order to reach the cytoplasm to be translated to functional protein. mRNA uptake mechanisms seem to be cell type dependent, and the physicochemical properties of the mRNA complexes can profoundly influence cellular delivery and organ distribution.
There are two basic approaches for the delivery of mRNA vaccines that have been described to date. First, loading of mRNA into DCs ex vivo, followed by re-infusion of the transfected cells58; and second, direct parenteral injection of mRNA with or without a carrier.
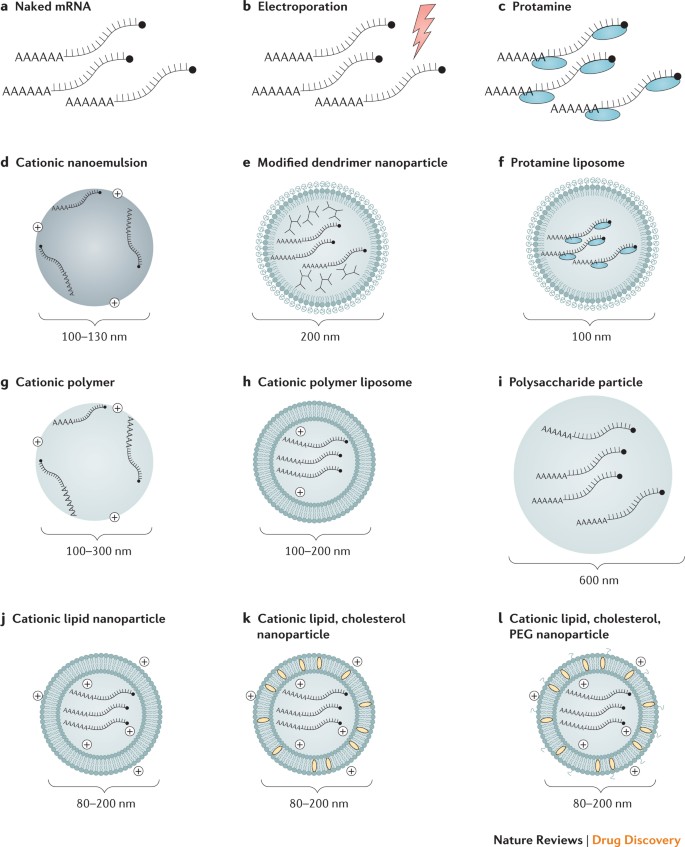
Ex vivo DC loading allows precise control of the cellular target, transfection efficiency and other cellular conditions, but as a form of cell therapy, it is an expensive and labour-intensive approach to vaccination. Direct injection of mRNA is comparatively rapid and cost-effective, but it does not yet allow precise and efficient cell-type-specific delivery, although there has been recent progress in this regard59. Both of these approaches have been explored in a variety of forms (Fig. 2; Table 1).
Ex vivo loading of DCs. DCs are the most potent antigen-presenting cells of the immune system. They initiate the adaptive immune response by internalizing and proteolytically processing antigens and presenting them to CD8+ and CD4+ T cells on major histocompatibility complexes (MHCs), namely, MHC class I and MHC class II, respectively. Additionally, DCs may present intact antigen to B cells to provoke an antibody response60. DCs are also highly amenable to mRNA transfection. For these reasons, DCs represent an attractive target for transfection by mRNA vaccines, both in vivo and ex vivo.
Although DCs have been shown to internalize naked mRNA through a variety of endocytic pathways61,62,63, ex vivo transfection efficiency is commonly increased using electroporation; in this case, mRNA molecules pass through membrane pores formed by a high-voltage pulse and directly enter the cytoplasm (reviewed in Ref. 64). This mRNA delivery approach has been favoured for its ability to generate high transfection efficiency without the need for a carrier molecule. DCs that are loaded with mRNA ex vivo are then re-infused into the autologous vaccine recipient to initiate the immune response. Most ex vivo-loaded DC vaccines elicit a predominantly cell-mediated immune response; thus, they have been used primarily to treat cancer (reviewed in Ref. 58).
Injection of naked mRNA in vivo. Naked mRNA has been used successfully for in vivo immunizations, particularly in formats that preferentially target antigen-presenting cells, as in intradermal61,65 and intranodal injections66,67,68. Notably, a recent report showed that repeated intranodal immunizations with naked, unmodified mRNA encoding tumour-associated neoantigens generated robust T cell responses and increased progression-free survival68 (discussed further in Box 2).
Physical delivery methods in vivo. To increase the efficiency of mRNA uptake in vivo, physical methods have occasionally been used to penetrate the cell membrane. An early report showed that mRNA complexed with gold particles could be expressed in tissues using a gene gun, a microprojectile method69. The gene gun was shown to be an efficient RNA delivery and vaccination method in mouse models70,71,72,73, but no efficacy data in large animals or humans are available. In vivo electroporation has also been used to increase uptake of therapeutic RNA74,75,76; however, in one study, electroporation increased the immunogenicity of only a self-amplifying RNA and not a non-replicating mRNA-based vaccine74. Physical methods can be limited by increased cell death and restricted access to target cells or tissues. Recently, the field has instead favoured the use of lipid or polymer-based nanoparticles as potent and versatile delivery vehicles.
Protamine. The cationic peptide protamine has been shown to protect mRNA from degradation by serum RNases77; however, protamine-complexed mRNA alone demonstrated limited protein expression and efficacy in a cancer vaccine model, possibly owing to an overly tight association between protamine and mRNA36,78. This issue was resolved by developing the RNActive vaccine platform, in which protamine-formulated RNA serves only as an immune activator and not as an expression vector52.
Cationic lipid and polymer-based delivery. Highly efficient mRNA transfection reagents based on cationic lipids or polymers, such as TransIT-mRNA (Mirus Bio LLC) or Lipofectamine (Invitrogen), are commercially available and work well in many primary cells and cancer cell lines9,13, but they often show limited in vivo efficacy or a high level of toxicity (N.P. and D.W., unpublished observations). Great progress has been made in developing similarly designed complexing reagents for safe and effective in vivo use, and these are discussed in detail in several recent reviews10,11,79,80. Cationic lipids and polymers, including dendrimers, have become widely used tools for mRNA administration in the past few years. The mRNA field has clearly benefited from the substantial investment in in vivo small interfering RNA (siRNA) administration, where these delivery vehicles have been used for over a decade. Lipid nanoparticles (LNPs) have become one of the most appealing and commonly used mRNA delivery tools. LNPs often consist of four components: an ionizable cationic lipid, which promotes self-assembly into virus-sized (~100 nm) particles and allows endosomal release of mRNA to the cytoplasm; lipid-linked polyethylene glycol (PEG), which increases the half-life of formulations; cholesterol, a stabilizing agent; and naturally occurring phospholipids, which support lipid bilayer structure. Numerous studies have demonstrated efficient in vivo siRNA delivery by LNPs (reviewed in Ref. 81), but it has only recently been shown that LNPs are potent tools for in vivo delivery of self-amplifying RNA19 and conventional, non-replicating mRNA21. Systemically delivered mRNA–LNP complexes mainly target the liver owing to binding of apolipoprotein E and subsequent receptor-mediated uptake by hepatocytes82, and intradermal, intramuscular and subcutaneous administration have been shown to produce prolonged protein expression at the site of the injection21,22. The mechanisms of mRNA escape into the cytoplasm are incompletely understood, not only for artificial liposomes but also for naturally occurring exosomes83. Further research into this area will likely be of great benefit to the field of therapeutic RNA delivery.
The magnitude and duration of in vivo protein production from mRNA–LNP vaccines can be controlled in part by varying the route of administration. Intramuscular and intradermal delivery of mRNA–LNPs has been shown to result in more persistent protein expression than systemic delivery routes: in one experiment, the half-life of mRNA-encoded firefly luciferase was roughly threefold longer after intradermal injection than after intravenous delivery21. These kinetics of mRNA–LNP expression may be favourable for inducing immune responses. A recent study demonstrated that sustained antigen availability during vaccination was a driver of high antibody titres and germinal centre (GC) B cell and T follicular helper (TFH) cell responses84. This process was potentially a contributing factor to the potency of recently described nucleoside-modified mRNA–LNP vaccines delivered by the intramuscular and intradermal routes20,22,85. Indeed, TFH cells have been identified as a critical population of immune cells that vaccines must activate in order to generate potent and long-lived neutralizing antibody responses, particularly against viruses that evade humoral immunity86. The dynamics of the GC reaction and the differentiation of TFH cells are incompletely understood, and progress in these areas would undoubtedly be fruitful for future vaccine design (Box 3).
*-*-*-*-*-
Box 2: Personalized neoepitope cancer vaccines
Sahin and colleagues have pioneered the use of individualized neoepitope mRNA cancer vaccines121. They use high-throughput sequencing to identify every unique somatic mutation of an individual patient’s tumour sample, termed the mutanome. This enables the rational design of neoepitope cancer vaccines in a patient-specific manner, and has the advantage of targeting non-self antigen specificities that should not be eliminated by central tolerance mechanisms. Proof of concept has been recently provided: Kreiter and colleagues found that a substantial portion of non-synonymous cancer mutations were immunogenic when delivered by mRNA and were mainly recognized by CD4+ T cells176. On the basis of these data, they generated a computational method to predict major histocompatibility complex (MHC) class II-restricted neoepitopes that can be used as vaccine immunogens. mRNA vaccines encoding such neoepitopes have controlled tumour growth in B16-F10 melanoma and CT26 colon cancer mouse models. In a recent clinical trial, Sahin and colleagues developed personalized neoepitope-based mRNA vaccines for 13 patients with metastatic melanoma, a cancer known for its high frequency of somatic mutations and thus neoepitopes. They immunized against ten neoepitopes per individual by injecting naked mRNA intranodally. CD4+ T cell responses were detected against the majority of the neoepitopes, and a low frequency of metastatic disease was observed after several months of follow-up68. Interestingly, similar results were also obtained in a study of analogous design that used synthetic peptides as immunogens rather than mRNA177. Together, these recent trials suggest the potential utility of the personalized vaccine methodology.
*-*-*-*-*-
Box 3: The germinal centre and T follicular helper cells
The vast majority of potent antimicrobial vaccines elicit long-lived, protective antibody responses against the target pathogen. High-affinity antibodies are produced in specialized microanatomical sites within the B cell follicles of secondary lymphoid organs called germinal centres (GCs). B cell proliferation, somatic hypermutation and selection for high-affinity mutants occur in the GCs, and efficient T cell help is required for these processes178. Characterization of the relationship between GC B and T cells has been actively studied in recent years. The follicular homing receptor CXC-chemokine receptor 5 (CXCR5) was identified on GC B and T cells in the 1990s179,180, but the concept of a specific lineage of T follicular helper (TFH) cells was not proposed until 2000 (Refs 181, 182). The existence of the TFH lineage was confirmed in 2009 when the transcription factor specific for TFH cells, B cell lymphoma 6 protein (BCL-6), was identified183,184,185. TFH cells represent a specialized subset of CD4+ T cells that produce critical signals for B cell survival, proliferation and differentiation in addition to signals for isotype switching of antibodies and for the introduction of diversifying mutations into the immunoglobulin genes. The major cytokines produced by TFH cells are interleukin-4 (IL-4) and IL-21, which play a key role in driving the GC reaction. Other important markers and functional ligands expressed by TFH cells include CD40 ligand (CD40L), Src homology domain 2 (SH2) domain-containing protein 1A (SH2D1A), programmed cell death protein 1 (PD1) and inducible T cell co-stimulator (ICOS)186. The characterization of rare, broadly neutralizing antibodies to HIV-1 has revealed that unusually high rates of somatic hypermutation are a hallmark of protective antibody responses against HIV-1 (Ref. 187). As TFH cells play a key role in driving this process in GC reactions, the development of new adjuvants or vaccine platforms that can potently activate this cell type is urgently needed.
reference link : https://www.nature.com/articles/nrd.2017.243



{kind=link}