









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



In an in-depth study of how COVID-19 affects a patient’s brain, National Institutes of Health researchers consistently spotted hallmarks of damage caused by thinning and leaky brain blood vessels in tissue samples from patients who died shortly after contracting the disease.
In addition, they saw no signs of SARS-CoV-2 in the tissue samples, suggesting the damage was not caused by a direct viral attack on the brain. The results were published as a correspondence in the New England Journal of Medicine.
Our results suggest that this may be caused by the body’s inflammatory response to the virus” said Avindra Nath, M.D., clinical director at the NIH’s National Institute of Neurological Disorders and Stroke (NINDS) and the senior author of the study.
“We hope these results will help doctors understand the full spectrum of problems patients may suffer so that we can come up with better treatments.”
Although COVID-19 is primarily a respiratory disease, patients often experience neurological problems including headaches, delirium, cognitive dysfunction, dizziness, fatigue, and loss of the sense of smell. The disease may also cause patients to suffer strokes and other neuropathologies.
Several studies have shown that the disease can cause inflammation and blood vessel damage. In one of these studies, the researchers found evidence of small amounts of SARS-CoV-2 in some patients’ brains. Nevertheless, scientists are still trying to understand how the disease affects the brain.
In this study, the researchers conducted an in-depth examination of brain tissue samples from 19 patients who had died after experiencing COVID-19 between March and July 2020.
Samples from 16 of the patients were provided by the Office of the Chief Medical Examiner in New York City while the other 3 cases were provided by the department of pathology at the University of Iowa College of Medicine, Iowa City. The patients died at a wide range of ages, from 5 to 73 years old.
They died within a few hours to two months after reporting symptoms. Many patients had one or more risk factors, including diabetes, obesity, and cardiovascular disease. Eight of the patients were found dead at home or in public settings. Another three patients collapsed and died suddenly.
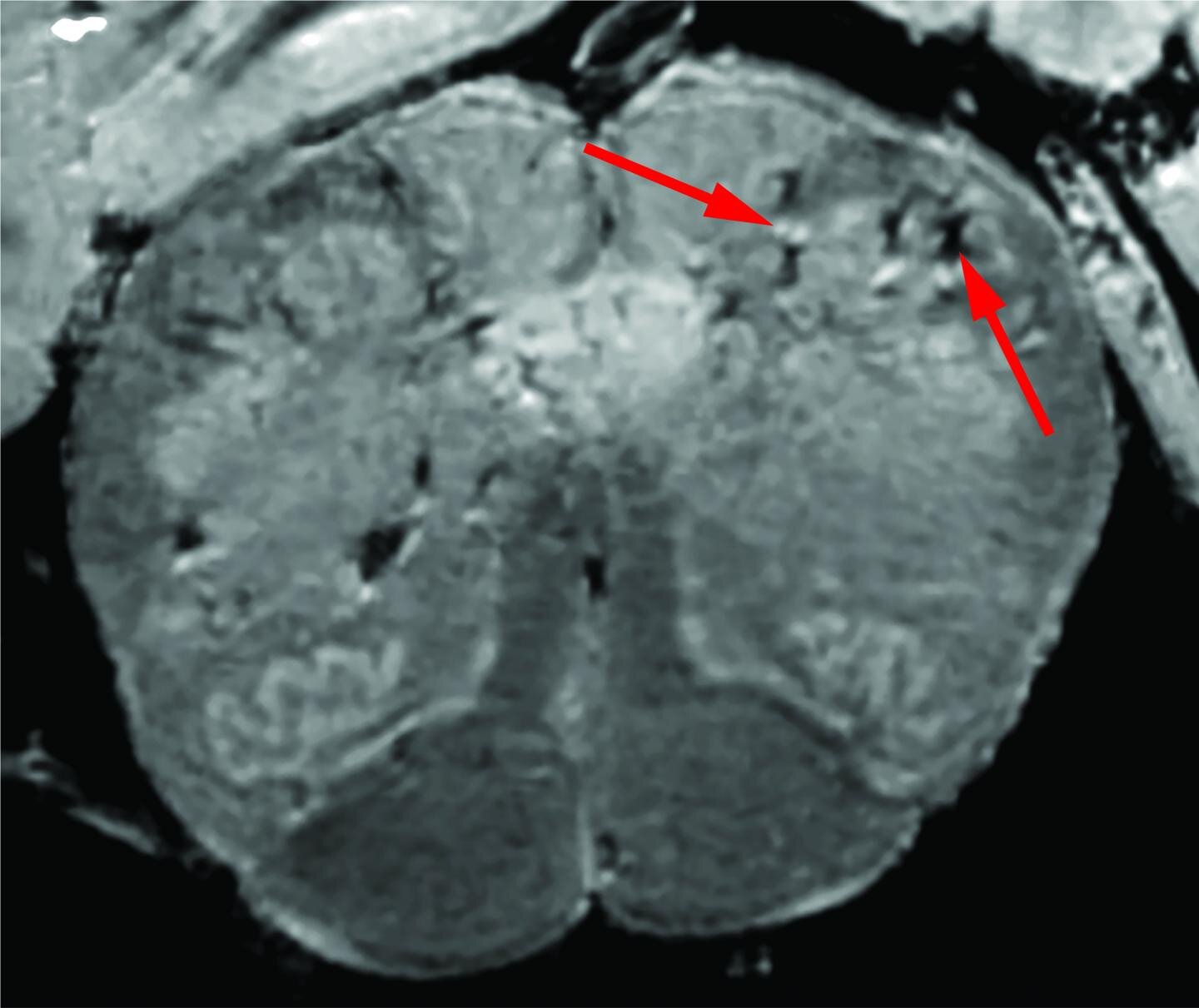
Initially, the researchers used a special, high-powered magnetic resonance imaging (MRI) scanner that is 4 to 10 times more sensitive than most MRI scanners, to examine samples of the olfactory bulbs and brainstems from each patient.
These regions are thought to be highly susceptible to COVID-19. Olfactory bulbs control our sense of smell while the brainstem controls our breathing and heart rate. The scans revealed that both regions had an abundance of bright spots, called hyperintensities, that often indicate inflammation, and dark spots, called hypointensities, that represent bleeding.
The researchers then used the scans as a guide to examine the spots more closely under a microscope. They found that the bright spots contained blood vessels that were thinner than normal and sometimes leaking blood proteins, like fibrinogen, into the brain.
This appeared to trigger an immune reaction. The spots were surrounded by T cells from the blood and the brain’s own immune cells called microglia. In contrast, the dark spots contained both clotted and leaky blood vessels but no immune response.
“We were completely surprised. Originally, we expected to see damage that is caused by a lack of oxygen. Instead, we saw multifocal areas of damage that is usually associated with strokes and neuroinflammatory diseases,” said Dr. Nath.
Finally, the researchers saw no signs of infection in the brain tissue samples even though they used several methods for detecting genetic material or proteins from SARS-CoV-2.
“So far, our results suggest that the damage we saw may not have been not caused by the SARS-CoV-2 virus directly infecting the brain,” said Dr. Nath.
“In the future, we plan to study how COVID-19 harms the brain’s blood vessels and whether that produces some of the short- and long-term symptoms we see in patients.”
Coronaviruses (CoVs) belong to the order Nidovirales and family Coronaviridae which is grouped on four genera based on phylogeny: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus (International Committee for Taxonomy of Virus; https://talk.ictvonline.org/).
CoVs are envelopment and spherical particles of 80 to 120 nm in diameter with a crown-like structures. This family contains the largest and non-segmented RNA genome, which is formed with a single-strand positive-sense around 27 to 32 kb in size [1]. CoVs’ genome shares structural organization, although differs in the base pairs number and sequence, even among closely related CoVs. The open reading frames 1a/b (ORF1a and ORF1b), located at the 5′ end encode non-structural components, the polyproteins pp1a and pp1b.
Two viral proteases cleave these proteins to generate 16 non-structural proteins (nsp1 to nsp16), including the RNA-dependent RNA polymerase (RdRP), an important protein involved in genome transcription and replication. The 3′ end includes ORFs that encode four major structural proteins: the spike surface glycoprotein (S), a small envelope protein (E), membrane protein (M), and nucleocapsid protein (N) that covers the RNA (Fig. (Fig.1).1). Besides, the CoV genome maintains genes that encode accessory proteins indispensable for adaptation and virulence and be successful to specific host [2, 3].

SARS-CoV-2 genome organization. The SARS-CoV-2 genome size is around 32 kb and is an RNA single-strand positive-sense that encodes 16 non-structural proteins (5′ end) and 4 structural proteins (3′ end) (S, E, M, and N) and 6 accessory proteins. SARS-CoV-2 genome. The genome contains a PoliA tail at 3′ end
The Betacoronavirus are zoonotic pathogens that have a wild animal origin, for example, bat or rodent origin. This group includes pathogenic human CoVs (HCoVs), such as HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1, that infect mammals and provoke mild related respiratory illness in infants, young children, elderly individuals, and immunocompetent hosts [1, 3].
However, the severe acute respiratory syndrome-related CoV (SARS-CoV) and the Middle East respiratory syndrome-related CoV (MERS-CoV) cause a severe respiratory syndrome in humans that aroused large-scale pandemics during 2002–2003 and 2012, respectively [4, 5]. Importantly, late in December 2019, Wuhan Municipal Health Commission, China, reported a cluster of cases of atypical pneumonia in Wuhan, Hubei Province, China, associated with a virus that rapidly spread all over the world.
Neurotropism of SARS-CoV-2
Up to date, many reports have described the association between respiratory viral infections with neurological symptoms. There are several recognized respiratory pathogens that gain access to the central nervous system (CNS), for instance, respiratory syncytial virus, the influenza virus, the human metapneumovirus, and HCoVs (HCoV229E, HCoV-OC43, and SARS-CoV) [39], that induce manifestations such as febrile or afebrile seizures, among other encephalopathies [40, 41].
Primary cultures of human astrocytes and microglia and various human neuronal cell lines, such as the neuroblastoma SK-N-SH, the neuroglioma H4, and the oligodendrocytic MO3.13, have potential tropism for HCoV-OC43 [42]. Using an experimental animal model, HCoV-OC-43 infection also showed neuro-invasiveness and neuro-virulence [43].
Therefore, it is not surprising to find brain SARS-positive autopsies. Using in situ hybridization, the SARS genomic sequence has been detected in the cytoplasm of neurons of the hypothalamus and cerebral cortex [44]. Furthermore, Moriguchi et al. [45] confirmed the presence of the new SARS-CoV-2 in cerebral spinal fluid.
In accord, epidemiological and clinical research have described neurological, non-common symptoms, and neurological manifestations associated with the SARS-CoV-2 infection (Table (Table2).2). These clinical features include neuralgia, confusion, hyposmia, hypogeusia, and altered consciousness, symptoms that evidence the neurotropic invasion by SARS-CoV-2 [19, 41].
Table 2
Neurological manifestations associated with SARS-CoV-2 infection
| Type of study and data of patients | Neurological diagnostic, symptoms, and clinical specimen for SARS-CoV-2 detection | Reference |
|---|---|---|
| Case seriesn = 4 patients73 Y/A male, 83 Y/A female, 80 Y/A female, and 88 Y/A female | Acute strokeAltered mental status, facial droop, slurred speech, left-side weakness, hemiplegia, and aphasiaNot specific specimen | [46] |
| Case reportn = 2 patients31 Y/A male, 62 Y/A female | Hunt and Hess grade 3 subarachnoid hemorrhage from a rupture aneurysmHeadache and loss of consciousnessIschemic strokeNasal specimen | [47] |
| Case reportn = 5 patients,< 50 Y/A | Large-vessel strokeHeadache, dysarthria, numbness, hemiplegia, and reduced level of consciousnessNot specific specimen | [48] |
| Case reportn = 1 patient41 Y/A | MeningoencephalitisSeizure, lethargic, photophobia, worsening encephalopathy, disorientation, hallucinations, and neck stiffnessNot specified specimen | [49] |
| Case reportn = 1 patient24 Y/A | Meningitis/encephalitisfatigue and fever, vomit, seizures, unconsciousness, and neck stiffnessCerebral spinal fluid specimen | [45] |
| Retrospectively report n = 24 males and 19 females16–85 Y/A | Stroke and stroke with pulmonary thromboembolismGuillain-Barré syndromeNasopharyngeal specimen | [37] |
| Case reportn = 2 patients52 Y/A male, 39 Y/A male | Variants of Guillain-Barré syndromeMiller Fisher syndromeDiplopia, gait instability, headache, anosmia, and ageusiaPolyneuritis cranialis and ageusiaOropharyngeal specimen | [50] |
| Case reportn = 1 patient61 Y/A female | Acute Guillain-Barré syndromeLegs weakness and severe fatigueOropharyngeal specimen | [51] |
| Case reportn = 1 patient65 Y/A male | Guillain-Barré syndromeAcute progressive symmetric ascending quadriparesis, facial paresis bilaterallyOropharyngeal specimen | [52] |
| Case reportn = 5 patients | Guillain-Barré syndromeNasopharyngeal specimen | [53] |
| Case reportn = 1 patient | Guillain-Barré syndromeNasopharyngeal specimen | [54] |
| Case reportn = 1 patient58 Y/A female | Acute hemorrhagic necrotizing encephalopathyAltered mental statusNasopharyngeal specimen | [55] |
Despite the evidence demonstrating the neurotropism of respiratory viruses, the exact mechanism of neuro-invasion accomplished by viruses remains currently unknown. However, the route of invasion of the CNS has recently been described for HCoV-OC-43. This virus gains access to the CNS through the olfactory bulb, moving along the olfactory nerve.
Then, neuro-propagation occurs along the multiple axonal connections expanding through the CNS (e.g., neuron-to-neuron propagation or diffusing particles) [56]. Similar to HCoV-OC-43, a model in vivo of SARS-CoV infection suggested that the virus enters the brain via the olfactory bulb, and then, a transneuronal spread could occurs [57].
Also, some infectious blood-borne viruses primarily targeting peripheral organs have evolved strategies to thwart the blood-brain barrier (BBB). These strategies include direct infection of the brain microvascular endothelial cells that form the BBB, a paracellular entry that involves alteration of the tight junctions, or the “Trojan horse” invasion, via the traffic of infected monocytes/macrophages migrating across the BBB, in a similar manner as the not-respiratory immunodeficiency virus 1 (HIV-1) [58, 59].
Likewise, when human primary monocytes are activated following infection by HCoV-229E and eventually become macrophages, it can invade tissues, including the CNS [60, 61]. Additionally, it has been reported that through activation of the brain microendothelium, the damage caused by the inflammatory response, allows the virus to reach the CNS.
In this sense, the neuro-invasion of SARS-CoV-2 could occur through trans-synaptic transfer, via the olfactory nerve, infection of vascular endothelium, or leukocyte migration across the BBB [38].
SARS-CoV-2 and the Function of the BBB
The microvascular endothelial cells that form the BBB protect the CNS from a wide variety of toxins and microorganisms found in the blood. These cells express tight junction proteins that limit the movement between adjacent cells and are through specific transporters and receptor proteins that control entry and exit of molecules coming from the blood toward the brain parenchyma [84].
Therefore, the study of the damage induced to microvascular endothelial cells represents the central framework for understanding the molecular mechanisms of virus infection in the CNS [85, 86].
Disruption of the BBB occurs upon infection with several recognized neurotropic viruses. Arbovirus that belongs to the Flaviviridae family, such as the West Nile Virus and the Zika virus, can induce damage in the BBB caused by the host cell’s response to viral factors.
Experiments carried out using in vitro and in vivo models of the BBB have demonstrated that these viruses replicate in the brain microvascular endothelial cells and induce down-regulation and degradation of tight junction proteins leading to disruption of the BBB [87–90]. Similarly, Bleau et al. [91] evaluated the ability of CoV to enter the CNS, using the highly hepatotropic mouse hepatitis virus type 3 and the weakly hepatotropic mouse hepatitis type A59.
The type 3-infected mice showed brain invasion that correlated with enhanced BBB permeability. The effect was associated with decreased expression of the zona occludens protein 1, the VE-cadherin, and the occludin. Since CoV are molecularly related in its mode of replication, it is speculated that other types of CoV use a similar mechanism of action to infect the brain microvascular endothelial cells [92, 93].
Importantly, it has been identified the presence of SARS-CoV-2 in the brain microvascular endothelial cells in frontal lobe tissue obtained at postmortem examination from a patient with COVID-19 [94]. Besides, viral particles and viral genome sequences of SARS-CoV have been detected in the cytoplasm of neurons of the brain, mainly in the hypothalamus and the cortex [44, 95]. This evidence suggests that SARS-CoV-2 crosses the BBB as well as others HCoV.
Therefore, infection by several respiratory viruses, including SARS-CoV-2, affects the integrity of the BBB through different mechanisms. The virus causes direct cell stress, associated with most of the cytotoxic effects that lead to degeneration of infected cells, for example, SARS-CoV induces apoptosis [96].
Endothelial cells activation as part of the inflammatory response causes an increase in the expression of proteases, such as matrix metalloproteinase, that promotes the degradation of the tight junction proteins [97]. However, it is probable that the inflammatory response plays the most important role in the induction of the damage to BBB.
The Inflammatory Response
The regular activity of neuroinflammation is mainly to restore the homeostasis in the brain [98]. However, prolonged CNS inflammation and systemic inflammatory response as a result of a wide variety of pathologies such as viral infections may influence the BBB integrity and further outcome in neurological disorders [99, 100]. Therefore, SARS-CoV-2 could cause damage to the BBB through the activation of the inflammatory immune response associated with a dysregulation around this process [101].
Activation of the microvascular endothelial cells has been associated with changes in BBB permeability. For example, during physiological conditions, immune cell migration into the CNS is rigorously controlled by mechanisms that operate at the level of the BBB.
Notably, migration of circulating immune cells into the CNS is low and restricted to specific innate and adaptative immune cell subsets, such as lymphocytes, macrophages, and antigen presenting cells as dendritic cells that maintain immune surveillance in the CNS [102].
However, during viral infections, the migration of immune cells is increased. This is supported by histopathologic examination of the brain tissue in patients with SARS-CoV, where pathological infiltration of CD68+ monocytes/macrophages and CD3+ T lymphocytes has been found in the brain mesenchyme [95].
Similarly, the infiltration process, related to interactions between the β1 and β2 integrins expressed on leukocytes and their ligands [i.e., intercellular adhesion molecules (ICAM): ICAM-1, ICAM-2, and vascular cell adhesion molecule-1 (VCAM-1)] present on the surface of the microendothelial cells, that induce extravasation across the BBB under inflammatory conditions has been reported [103–107].
Evidence suggests that infection and activation of the microvascular endothelial cells by typical neurotrophic viruses increased endothelial adhesion molecules expression [88]. This condition facilitates the trafficking of viruses-infected immune cells into the CNS via the ‘Trojan horse’ mechanism [88].
Likewise, during viral replication in the host cells, the damage is caused because SARS-CoV-2 is a cytopathic virus that induces the release of damage-associated molecular patterns (DAMPs) [108]. DAMPs are endogenous molecules released from damaged cells that interact with molecules called pattern-recognition receptor (PRR) that induce in the neighboring epithelial cells, endothelial cells, and macrophages a state of high inflammation [109].
Once the virus interacts with the host cells, the viral genome and viral proteins can also be recognized by PRRs and activated the immune response. Different PRRs recognize SARS-CoV-2, for example, the Toll-like receptors (TLR), which are molecules expressed in many cell lines, including endothelial cells, macrophages, and dendritic cells. TLR3, TLR7, TLR8, and TLR9 induce several pathways of activation that produce proinflammatory cytokines and other antiviral molecules to control the infection. However, this response can be dysregulated and exacerbated cytokines production [110].
Also, NOD-like receptor (NLR), other PRR, activates the inflammasome complex and induces the activation state in some cell types such as macrophages and epithelial and even in the microvascular endothelial cells leading to high production of interleukin (IL)-1β and IL-18. Nevertheless, this mechanism needs to be studied in detail for the new CoV [111].
On the other hand, the viral RNA activates typical molecules such as the retinoic acid inducible gene 1 and the melanoma differentiation associated gene 5 and induces an antiviral state, in which interferons (IFN) are secreted (mainly IFN type I and III). Interferons are molecules important to clearance the viral infection to prevent viruses from replicating [112, 113].
In patients with COVID-19, high levels of IFN, especially IFN I, are detected; this molecule blocks the viral replication in adjacent cells and produces some effects against the viral infection such as the induction of interferon-stimulated gene expression, the stimulation of cytokines production, and the activation of immune response cells (i.e., macrophages, monocytes, and neutrophils) [112, 114]. Other CoV infections have a similar response [115, 116].
Furthermore, patients infected with SARS-CoV-2 have increased levels of several cytokines and chemokines: TNF-α, IFN-γ, interleukin-1 receptor antagonist (IL-1RA), IL-2, IL-6, IL-7, IL-8, IL-9, IL-10, and the granulocyte macrophage colony-stimulating factor; importantly, high levels of IL-6 have been linked to a worse prognosis in COVID-19 patients [116, 117].
This high production and misbalance of all these molecules is defined as cytokines storm (CS), which could be an essential factor to cause disruption of the BBB [110]. Interestingly, CS induces the activation of platelets, neutrophils, monocytes, and macrophages; additionally, some of these molecules can interact with the complement and the coagulation systems and contribute to the pathogenic inflammation [117].
Furthermore, some chemokines can attract some innate immune response cells such as monocytes, natural killer cells, dendritic cells, and T cells [118] and induce the production others cytokines such as the monocyte chemotactic protein-1, the granulocyte colony-stimulating factor, the macrophages inflammatory protein 1-α, and IL-10 that recruit lymphocytes and monocytes and initiate the humoral response. Together, all these mechanisms can contribute to the severity of neurological symptoms of SARS-CoV-2 infection in the BBB [30].
Other physiological disturbances in COVID-19 patients such as thrombocytopenia, lymphopenia (CD8+ T, CD4+ T, Treg cells, and platelets), and eosinopenia have also been describing. Blood samples and spleen and lymph nodes present these types of dysregulation due to the recruited cells to the infected sites to control the viral replication (Table (Table3)3) [119, 120]. Also, several patients present high levels of D-dimer in the early stage of the infection (Table (Table3).3). This molecule is an important marker in the disorder of coagulation. It represents a thrombotic state that leads to embolic vascular events and can produce venous clots and induce brain damage [121]. Therefore, COVID-19 patients with high inflammation trigger excessive thrombin production that inhibits fibrinolysis and activates the complement pathways leading thromboinflammation, microthrombin deposition, and microvascular dysfunction associated with damage of the BBB [122, 123].
Table 3
Laboratory findings in neurological manifestation in COVID-19
| Author | Manifestation | Laboratory finding | Reference |
|---|---|---|---|
| Avula et al. | Stroke | – Lymphopenia | [46, 48] |
| – Elevated C-reactive [26 mg/dl (0.04 mg/dl)] | |||
| – Elevated D-dimer [mean 8704 ng/ml (< 880 ng/ml) | |||
| – Elevated lactate dehydrogenase (712 U/L) | |||
| Oxley et al. | – D-dimer [5972 ng/ml (0–500 ng/ml)] | ||
| Moringuchi et al. | Meningoencephalitis | – Elevated neutrophil | [45] |
| – Increased C-reactive protein | |||
| Guitierrez-Ortiz et al. | Guillain-Barré syndrome | – Lymphopenia (1000 cells/μl) | [50, 51, 54] |
| – Leucopenia (3100/cells/μl) | |||
| – Elevated C-reactive protein (2.8 mg/dl) | |||
| – Positive GD1b-IgG ganglioside antibody | |||
| Virani et al. | – Lymphopenia and thrombocytopenia | ||
| Zhao et al. | – Lymphocytopenia [0.52 × 109/L (1.1–3.2 × 109/L)] | ||
| – Thrombocytopenia [113 × 109/L (125–3000 × 109/L)] |
An unexplored mechanism that could produce damage in the BBB is the adaptative immune response. The generation of antibodies (Abs) against SARS-CoV-2 can cross-react with some molecules of the brain microvascular endothelial cells and produce damage through the activation of the complement system (C3 and C4 proteins).
Also, Ab-dependent enhancement phenomenon can increase the infection and contribute to the injury. This process has been extensively studied in Dengue and Zika virus infection, where the Abs produced in the first exposure can cross-react in a second exposure and enhance the infection instead of neutralizing it [124, 125]. Additionally, the Abs can generate an autoimmune attack and interact with the virus forming immune complexes and induce the complement system activation [126].
Recently, some studies showed that the cellular immune response could be central to determine the disease condition. Several viruses, including SARS-CoV-2, can activate CD4+ and CD8+ and induce clonal expansion, specific cell effectors, and cellular memory [127, 128].
Also, T cells can cross-react inducing a state of protection observed in unexposed people with SARS-CoV-2 [129, 130]. Finally, more studies around the interaction with SARS-CoV-2 and the host immune system need to be clarified. Research around the mechanisms involved in inflammation response can allow the development of strategies that might help to mitigate the health consequence of this pandemic.
Neurological Implications of BBB Disruption by SARS-CoV-2
As previously discussed, multiple respiratory viruses can affect the CNS. For example, the mouse hepatitis virus induces inflammation, BBB damage, and demyelination in rat models [131]. Likewise, a case report of HCoV-OC43 detected in nasopharyngeal and cerebral spinal fluid samples from a child patient exhibited acute disseminated encephalomyelitis, a low-prevalence CNS disease that induces demyelination [132].
The H1N1 virus, the causative agent of high mortality rates, also presented neurological complications. A retrospective study of the clinical files of 55 patients infected with H1N1 detected 50% of visible neurological symptoms [133]. Interestingly, most patients with neurological manifestations due to H1N1 infection manifested brain edema [134].
Importantly, in autopsy studies, patients with SARS-CoV showed endothelial activation associated with the loss of cerebral vascular integrity displaying multifocal hemorrhage [135]. Histological examination of brain tissue specimens of patients with SARS-CoV infection also showed neuronal degeneration, necrosis, edema, extensive glial cell hyperplasia, and cellular infiltration of the vascular walls by monocytes and lymphocytes [40].
With this background, several studies have attempted to characterize the neurological manifestations of SARS-CoV-2. An increasing number of individual case reports have emerged describing acute neurological disorders ranging from Guillain-Barré syndrome and acute myelitis to acute hemorrhagic necrotizing encephalopathy [136]. Although the long-term neurological implications of SARS-CoV-2 infection are still unknown, important clues suggest that complications of the disease are related to CNS invasion by damaging the BBB.
Neurological Implications of BBB Disruption by SARS-CoV-2 in Long-Term Dementia
There is a very complex interaction between the brain cells and the cerebral vasculature. Consequently, preservation of the cerebrovascular function and its integrity has a central role in this sophisticated communication. Additionally, any derangements can have deleterious acute and chronic consequences such as the development of neurodegenerative diseases and dementia [137].
Infectious agents have been suspected as contributing factors to dementia, especially in Alzheimer’s disease (AD). Interestingly, the BBB disruption appears to be an early feature of this disease [138]. For instance, Bell et al. [139] demonstrated that BBB breakdown was derived in neurotoxic proteins infiltration (e.g., amyloid-β peptides, the hallmark of AD), affecting neurons and either initiating or exacerbating neurodegeneration.
Also, Ueno et al. [140] using experimental animal models exhibiting some phenotypes of vascular dementia showed that BBB damage might be related to amyloid-β peptides accumulation. Accordingly, damage induced to the BBB by infectious agents might trigger neurodegenerative diseases in predisposed patients [141]. Therefore, viral infections such as SARS-CoV-2 could be associated with an increased risk of AD development and a faster rate of cognitive decline in older populations.
AD in systemic virus infection is an example of a condition that is primarily neurodegenerative; however, in many cases, it is not clear whether BBB changes are the cause or the effect of a neuropathology. Furthermore, it is possible that the BBB anomalies and the disease drive each other in a self-perpetuating manner, contributing to damage progression [142]. As mentioned, acute and chronic systemic inflammation accelerates the progress of AD [143].
Also, a 5-year follow-up study showed that viral infections, like the induced by cytomegalovirus, are linked with faster cognitive decline and development of AD [144]. Furthermore, systemic inflammation in AD is associated with several BBB changes, which further favor amyloid-β peptides accumulation into the brain because the injury alters the influx and efflux of the peptides [145]; correspondingly, systemic inflammation accelerates hippocampal amyloid-β peptides deposition [146].
Therefore, dysfunction of the BBB might play a significant role in the pathogenesis of vascular dementia induced by SARS-CoV-2 infection, but further observations are needed.
Additionally, damage to the BBB is not the only mechanism where SARS-CoV-2 infection can result in dementia. Some data suggest that amyloid-β protein possesses antimicrobial and antiviral activity in vitro [147]. Therefore, the presence of insoluble deposits of amyloid-β peptides could be a factor (e.g., genetic predisposing) that alter the response to viral SARS-CoV-2 infection.
Thus, it is conceivable that SARS-CoV-2 contributes to BBB damage and also creates a feed-forward effect whereby pathogen-induced damage favors a further spread of the pathogen’s transit zones and even the sequential development of the pathology associated to AD [138].
On the other hand, individuals with AD are more vulnerable to the effects of peripheral infection, especially SARS-CoV-2, mainly due to the association of physical comorbidities. It is more probable that these individuals have cardiovascular disease, diabetes, and pneumonia [148]. Besides, there is an overall decrease in naive T cell diversity after the age of 65 [149–151]; this can limit the capacity of the individual to induce a sufficient immune response to infection. Together, these data indicate the vulnerability of these patients to infection.
Neurological Implications of BBB Disruption by SARS-CoV-2 in Multiple Sclerosis
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS, characterized by several pathological processes, including inflammation, trans-endothelial migration, demyelination, axonopathy, and neuron loss mediated by immune cells [152]. MS represents a neurological disease where an infectious agent plays a triggering role, being viruses the most likely culprit in genetically predisposed individuals [141].
There is a presumption that several neurotropic viruses using similar mechanisms could be involved in MS pathogenesis [153]. Some viruses that have been implicated in the development of MS include herpes viruses, paramyxoviruses, picornaviruses, as well as viruses that classically affect the respiratory system such as influenza virus [154].
A critical step in the pathogenesis of MS is the infiltration of autoreactive CD4+ T-lymphocytes into the CNS after activation in the periphery. Evaluation of proinflammatory cytokines (IL-1β, IL-6, and IL-8), Th1 cytokines (IFN-γ, TNF-α, IL-2, and IL-12), and Th2 cytokines (IL-4, IL-5, and IL-10) in sera collected from SARS patients within 2 days of hospital admission showed a substantial elevation of IL-12, IL-6, IL-8, IL-10, and IFN-γ [155].
Also, Sonar et al. [156] revealed that IFN-γ favored the trans-endothelial migration of CD4+ T cells from the apical (luminal side) to the basal side (abluminal side) of the endothelial monolayer (Fig. (Fig.2).2). Besides, using multicolor immunofluorescence and confocal microscopic analysis, these authors indicated that IFN-γ induce relocalization of ICAM-1, platelet endothelial cell adhesion molecule-1, zona occludens protein 1, and VE-cadherin in the endothelial cells.
These findings reveal that the IFN-γ produced during the response to infection and inflammation could contribute to the disruption of the BBB and promote CD4+ T cells brain migration. Interestingly, BBB disruption appears in experimental autoimmune encephalomyelitis, a typical MS model, and the clinical severity is linked to the degree of BBB integrity [157]. Furthermore, imaging studies showed BBB disruption in normal-appearing white matter in MS [158].
This data is important since BBB breakdown precedes the development of new MS lesions [159]. In summary, it is possible that the damage caused by SARS-CoV-2 infection to the endothelial cells also causes loss of the BBB integrity, favoring MS progression.

Possible mechanism of damage to the blood-brain barrier (BBB) by the action of SARS-CoV-2. a Expression of angiotensin-converting enzyme 2 (ACE2) and the pro-protein convertase furin (PCF) in the membrane of the brain microvascular endothelial cells facilitates SARS-CoV-2 infection. b SARS-CoV2 infection activates the brain microendothelial cells inducing high expression of the vascular and the intercellular adhesion molecules (VCAM and ICAM). Likewise, SARS-CoV-2 induces the expression and activation matrix metalloproteinases (MMP) that degrade tight junctions proteins. c Recognition of ICAM and ICAM through the β1 and β2 integrins causes binding of circulating leukocytes to endothelial cells that lead transcellular extravasation. This process facilitate viral entrance to the cerebral parenchyma through the “Trojan horse” mechanism. d SARS-CoV-2 viral replication induces endothelial cell contraction and lysis. Increased permeability of the BBB allows extravasation of plasma proteins and blood cells. Activation of leukocytes and platelets contributes to the BBB damage. Besides, endothelial cell death disturbs the microenvironment of the brain parenchyma allowing free passage of the SARS-CoV-2 virus and infection of other cells of the central nervous system
reference link : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7518400/
More information: Myoung-Hwa Lee et al, Microvascular Injury in the Brains of Patients with Covid-19, New England Journal of Medicine (2020). DOI: 10.1056/NEJMc2033369



{kind=link}