











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A new study finds older adults who received positive airway pressure therapy prescribed for obstructive sleep apnea may be less likely to develop Alzheimer’s disease and other kinds of dementia.
Researchers from Michigan Medicine’s Sleep Disorders Centers analyzed Medicare claims of more than 50,000 Medicare beneficiaries ages 65 and older who had been diagnosed with OSA.
In this nationally representative study, they examined if those people who used positive airway pressure therapy were less likely to receive a new diagnosis of dementia or mild cognitive impairment over the next 3 years, compared to people who did not use positive airway pressure.
“We found a significant association between positive airway pressure use and lower risk of Alzheimer’s and other types of dementia over three years, suggesting that positive airway pressure may be protective against dementia risk in people with OSA,” says lead author Galit Levi Dunietz, Ph.D., M.P.H., an assistant professor of neurology and a sleep epidemiologist.
The findings stress the impact of sleep on cognitive function. “If a causal pathway exists between OSA treatment and dementia risk, as our findings suggest, diagnosis and effective treatment of OSA could play a key role in the cognitive health of older adults,” says study principal investigator Tiffany J. Braley, M.D., M.S., an associate professor of neurology.
Obstructive sleep apnea is a condition in which the upper airway collapses repeatedly throughout the night, preventing normal breathing during sleep. OSA is associated with a variety of other neurological and cardiovascular conditions, and many older adults are at high risk for OSA.
And dementia is also prevalent, with approximately 5.8 million Americans currently living with it, Braley says.
Obstructive sleep apnea (OSA) is a common breathing and sleeping disorder, the primary cause of which is aspiratory collapse of the pharyngeal airway, and leads to intermittent hypoxia (IH) (1). Previous studies have shown that OSA is associated with behavioral and neuropsychological deficits, including impaired spatial learning memory and cognition (2,3). However, the specific mechanisms underlying the chain of events from the development of IH to cognitive impairment remain elusive. It has been suggested that reactive oxygen species (ROS), which are produced in excess in cases of IH, are strongly associated with the presence of IH-induced cognitive impairment (4,5).
ROS are essential for many biological processes and these molecules are constantly formed in cells and removed by antioxidant defenses (6). Moreover, previous studies have demonstrated that increased oxidative stress is present in OSA (7-10). It has been shown that there is increased ROS production in stimulated neutrophils from patients with OSA, while the levels of ROS are attenuated after continuous positive airway pressure (CPAP) treatment (7,8). Furthermore, lipid peroxidation and protein carbonylation, both of which are markers of oxidative stress, are observed in patients with OSA (9,10). Moreover, these processes damage biomolecules such as lipids, proteins and DNA (6).
The aims of the present review were to examine the production of ROS and its role in the pathobiology of cognitive impairment in an OSA model and to investigate the underlying mechanism of ROS-induced cognitive deficiencies.
ROS formation
The ROS generated in cells include hydrogen peroxide (H2O2), oxygen ions (O2-), and hydroxyl and superoxide radicals (OH• and O2•-) (11). Although mitochondria are described as the major source of endogenous ROS generation, other subcellular compartments, including the cellular membrane, peroxisomes and the endoplasmic reticulum (ER), are involved in ROS generation and scavenging (12-15). This section of the review will focus on the ROS generation process in different subcellular compartments and its responses under IH conditions.
Mitochondria and ROS generation
As described above, the majority of ROS are generated from mitochondria during ATP synthesis, which supplies energy for physiological functions (Fig. 1A) (16). During this process, electrons flow along the mitochondrial electron transport chain (ETC) and protons are translocated from the mitochondrial matrix into the intermembrane space, thus creating a proton gradient (4). During normal aerobic respiration, approximately 2% of electrons ‘leak’ from the flow, resulting in the formation of O2•- (17). O2•- is easily detoxified by superoxide dismutase, which leads to the formation of H2O2 (18). While H2O2 is further reduced to H2O by glutathione peroxidase. H2O2 can also form highly reactive OH• by reacting with metal ions, which may be responsible for oxidative stress-induced cellular damage (19).
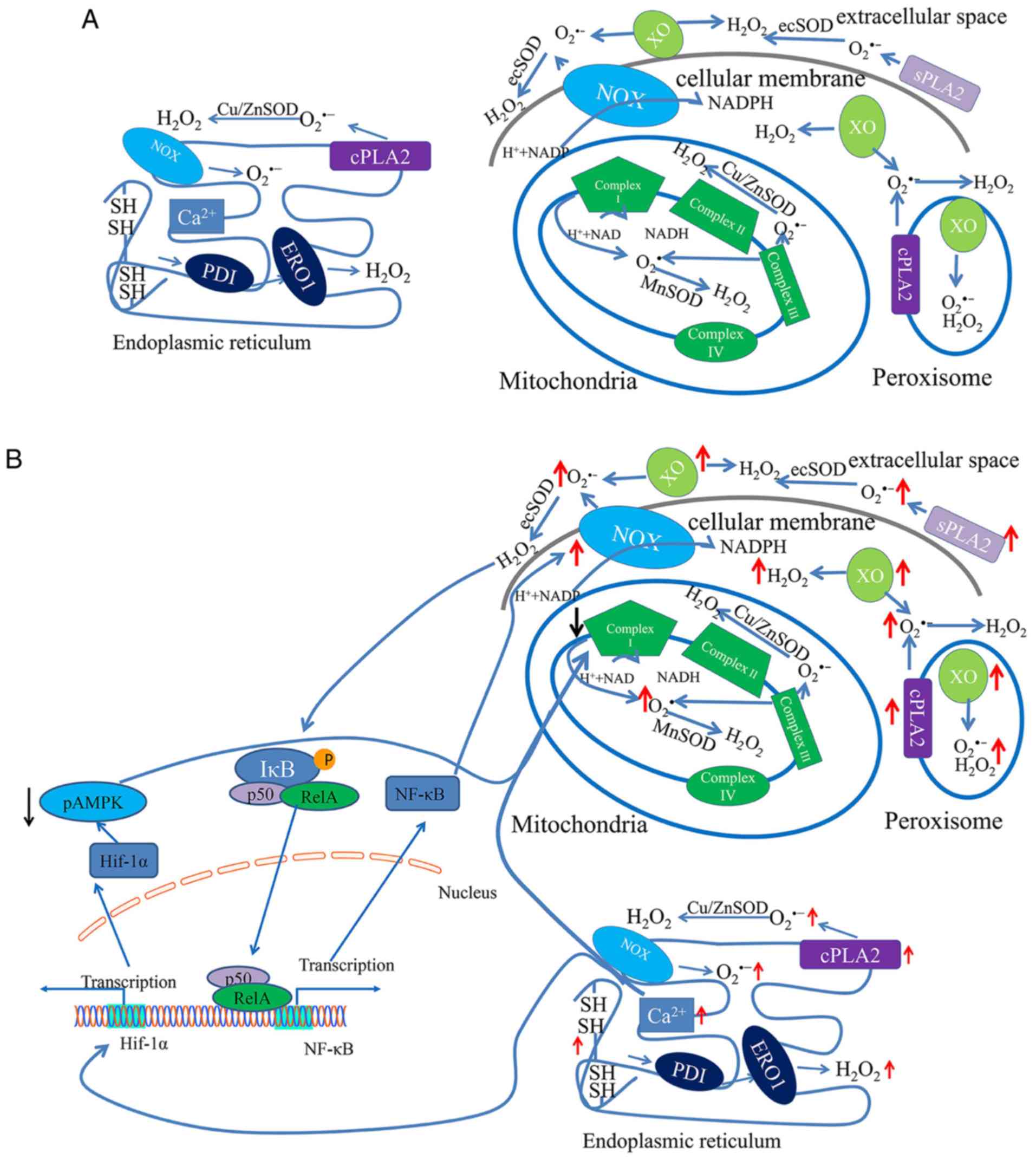
During mitochondrial respiration, some oxidoreductases affect ROS production, including complex I via complex IV (4). Under physiological conditions, a low concentration of ROS is maintained by the action of these oxidoreductases. However, increased ROS production can be caused by the inhibition of complex I activity in the mitochondrial ETC, under IH conditions (20). Furthermore, complex I, the first oxidoreductase in the ETC, is dependent on NADH-producing substrates to produce O2•- (21). A previous study demonstrated that IH induced complex I inhibition via the upregulation of NADPH oxidase (NOX) in PC12 cell cultures (22). Moreover, when complex I activity is decreased, ROS levels are increased in mitochondria due to the inability of electrons to be transported, which results in the formation of superoxide via a one-electron reduction of oxygen (23). However, complex III, another major producer of superoxide and ROS within the mitochondrial ETC, is unaffected (24). These results suggest that complex I, and not complex III, may be involved in increasing ROS levels (Fig. 1B).
ER and ROS generation
The ER is a large organelle that participates in the correct assembly and folding of nascent proteins and is also the site of post-translational protein modifications in ATP-dependent chaperone-mediated processes (25). Furthermore, approximately 25% of ROS are derived from the ER and are primarily required for oxidative protein folding (26). Correct protein folding involves the formation of disulfide bonds in a process driven by protein disulfide isomerase (PDI) and ER oxidoreductin (ERO-1) (27). In this process, electrons are transferred from PDI to O2, resulting in H2O2 formation (Fig. 1A) (27).
To perform these functions, the ER lumen possesses a unique environment composed of molecular chaperones, folding enzymes and high concentrations of ATP and calcium (28). In addition, the oxidative environment favors protein folding and, in particular, the formation of intra- and intermolecular disulfide bonds (29). However, increased ROS production may lead to a loss of ER homeostasis and the subsequent accumulation of misfolded proteins, a process known as ER stress (30). Under conditions of ER stress, additional misfolded or unfolded proteins are synthesized, leading to the depletion of glutathione (GSH) (31). After GSH is utilized, the oxidizing environment facilitates the reoxidation of protein thiols through interaction with ERO-1/PDI (27). These steps produce repetitive cycles of disulfide bond breakage and formation, with each cycle generating additional ROS as a byproduct (25). Furthermore, the accumulation of unfolded proteins in the ER elicits Ca2+ leakage into the cytosol, thus causing increased ROS production in the mitochondria (Fig. 1B) (29).
NOX
NOX is a multisubunit enzymatic complex that is localized to both cellular and subcellular membranes (32). Furthermore, NOX catalyzes O2•-production via the one-electron reduction of O2 using NADPH (33). There have been ≥7 NOX isoforms, including NOX1-5 and DUOX1-2, identified in this enzymatic system (34). Moreover, NOX can regulate electron flow and transfer to the outer heme, where the electron accepts O2 to form O2•- (35).
As a complex of major ROS-generating enzymes, NOX plays an important role in the OSA model (36). NOX2 expression and ROS production have been shown to be simultaneously increased in patients with OSA (37). Furthermore, pharmacological inhibition of NOX by apocynin and NOX2 deficiency can attenuate arterial hypertension in an OSA model by suppressing ROS production (38). In addition to cardiovascular risk, the cognitive deficits induced by oxidative stress are partially mediated by excessive NOX activity during IH (39). Furthermore, mice lacking NOX activity present with a learning ability comparable to that of IH-exposed wild-type littermates, which exhibit spatial learning deficits in a water maze test (40). Collectively, these studies indicated that NOX may be involved in ROS production in a model of OSA. However, the specific mechanistic involvement of NOX in IH injury requires further investigation.
Peroxisomes and ROS generation
In addition to mitochondria and the ER, superoxide and other ROS are generated in subcellular organelles peroxisomes (Fig. 1). The primary function of these particles is the oxidative degradation of long-chain fatty acids (41). Peroxisomes are spherical or oval shaped particles with a diameter of 0.2-1 mm that are surrounded by a single membrane (42). Moreover, peroxisomes in mammals harbor >100 enzymes and other proteins, some of which are associated with OSA or IH (43).
Xanthine oxidase (XO)
XO is localized to the outer surface of the cellular membrane, as well as in the cytosol and peroxisomes (44). XO catalyzes the oxidation of xanthine to uric acid at the site of flavin adenine dinucleotide, together with the reduction of NAD+ and O2 (45). In this process, the affinity for O2 (44) is significantly enhanced, resulting in univalent and divalent electron transfer to O2 to generate O2•- and H2O2, respectively (46).
XO is a critical source of ROS in ischemia/reperfusion injury, which is similar to chronic cycles of hypoxia and reoxygenation (47). Moreover, IH processes are associated with elevated XO activity and promotion of ROS formation by increasing the proteolytic conversion of xanthine dehydrogenase to XO (48). XO has been identified to be involved in ROS production in OSA, as lipid peroxidation, a marker of oxidative stress, is reduced after the application of the XO inhibitor allopurinol (49). Furthermore, allopurinol reduces the level of ROS by decreasing free radical generation via inhibition of the XO system (50). These findings suggest that XO plays a key role in increased ROS production under IH conditions.
Nitric oxide (NO) synthase
NO is known to play a critical role in the regulation of cellular processes that include catalyzation by nitric oxide synthase (NOS) (51). Although excess NO can induce apoptosis via the aggravation of oxidative stress (52), the specific mechanism is not fully understood.
Moreover, two NOS enzymes, the constitutive calcium/calmodulin-dependent neuronal and endothelial isoforms (53), show sustained expression in the central nervous system. However, under certain pathological conditions, including ischemia, hypoxia and other pathological stimuli, another NOS, inducible calcium-independent isoform (iNOS), is activated (54). Furthermore, IH has been shown to augment NO generation by activating iNOS (55), which induces apoptosis via the aggravation of oxidative stress. In addition, overexpression of O2•-in the endothelial tissue of patients with OSA is reduced by the NOS inhibitor l-nitroarginine methyl-ester (56). NOS has also been revealed to play a critical role in enhancing oxidative stress in OSA (56).
Phospholipases A2 (PLA2s)
PLA2s belong to a family of enzymes that hydrolyze the acyl bond at the sn-2 position of phospholipids to generate free fatty acids and lysophospholipids (57). PLA2s can be classified into three families based on their calcium requirement for catalytic activity, including calcium-dependent cytosolic PLA2 (cPLA2), calcium-independent PLA2 and secretory PLA2 (sPLA2) (58). It has been shown that all these PLA2sare involved in ROS production via arachidonic acid and lipoxygenase, which produce O2•- (59).
While there is no direct evidence to establish a connection between PLA2 expression and IH treatment, PLA2 activity is significantly increased after ischemia/reperfusion, which is dependent on Ca2+ concentration (60). Previous studies have revealed that cPLA2 immunoreactivity is selectively higher in the hippocampal CA1 region compared with other regions in a hypoxia-ischemia rat model (61). Furthermore, inhibition of cPLA2 attenuates oxygen-glucose deprivation-induced neuronal death in the hippocampus, which suggests that cPLA2 is involved in ischemic injury (62). In addition, a biphasic increase in them RNA expression of sPLA2 in the cortex of a rat brain after 20 min of transient forebrain occlusion has been shown (63). These results suggest that enhanced PLA2 activity results in the increased production of ROS in ischemia/reperfusion. Thus, it is hypothesized that the excessive ROS levels under IH conditions may also be due to increased PLA2 activity.
Misregulation of ROS-sensitive transcription factors and their downstream genes, such as hypoxia-inducible factor (HIF)-1 α and NF-κB (10), also aggravates oxidative stress (Figs. 1B and 2).
Although many organelles and peroxisomes are involved in ROS production during IH treatment, there is no direct evidence that demonstrates the underlying mechanism responsible for ROS production (64). Therefore, on the basis of previous studies, it is hypothesized that mitochondrial dysfunction may be the main source of excessive ROS production in OSA (20,22,65). Thus, attenuation of mitochondrial dysfunction may be an effective strategy for future clinical therapeutics.
ROS-induced consequences lead to cognitive impairment in sleep apnea
OSA is commonly associated with cognitive impairment, particularly memory, verbal fluency, attention and perception impairments (66,67), but the cause of this cognitive dysfunction is not fully understood.
Various different mechanisms that link OSA to cognitive dysfunction have been suggested (4,5,68-70). A major proposed mechanism is IH, which induces a reduction in memory performance in patients with OSA via oxidative stress injury (71). Moreover, increased levels of ROS have been confirmed to contribute to learning and memory impairments (72). In addition, increased ROS production induced by IH can result in deficits in spatial learning and memory in rodent models, which are largely dependent on the hippocampus. Neuroimaging studies have been performed to investigate the effects of OSA (73-75). Regional gray matter (GM), which is closely associated with memory formation, is observed to be lost in multiple brain regions of patients with OSA via MRI of the cortex (75). In another study, voxel-based morphometry was performed to measure cortical GM volume and indicated that, compared with healthy control individuals, patients with OSA exhibited significant cognitive impairment and had a smaller right hippocampus (76). Therefore, the cortex and hippocampus are the two major regions associated with cognitive impairment in OSA models. Although the underlying mechanism is not fully understood, it is hypothesized that the density of capillaries and large-diameter vessels is abundant in the two regions and that these are sensitive to oxidative stress (77).
Many pathways, including mitochondrial dysfunction, inflammation, apoptosis, ER stress and neuronal activity disturbance, are proposed to be associated with cognitive impairment induced by oxidative stress (Fig. 2).
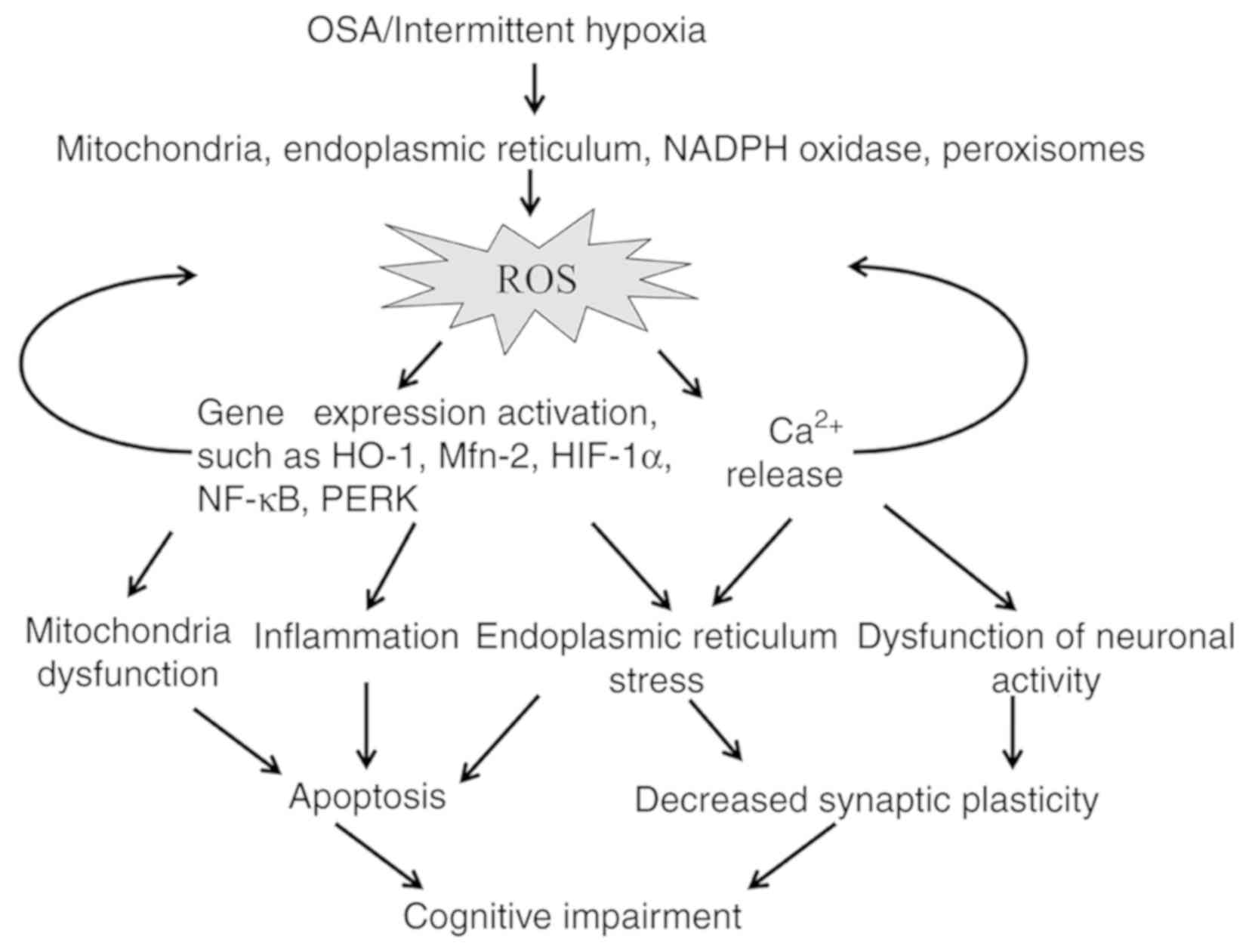
Mitochondrial dysfunction
Mitochondrial dysfunction is a common feature of patients with OSA, and the function of oxidoreductases in the mitochondrial ETC is disturbed under IH conditions (4). Furthermore, mitochondrial fusion and fission, which play important roles in mitochondrial function, have been observed in an IH model (78). Under IH conditions, mitochondrial fission occurs more frequently than fusion and leads to apoptosis through regulation of the expression of mitochondrial fusion protein-2 (Mfn2) (76).
Thus, ROS may control the expression of Mfn2. While the full mechanism has not been elucidated, it is hypothesized that the mechanism could be associated with the overexpression of heme oxygenase-1, a gene that responds to oxidative stress (79). In addition, a significant correlation is found between OSA severity and decreased mitochondrial DNA (mtDNA) copy number, suggesting that patients with a high apnea-hypopnea index are exposed to greater systemic blood oxidative stress (65). Despite the lack of direct evidence to show that mitochondrial dysfunction is a cause of cognitive impairment in OSA, clinical research has identified that the number of copies of mtDNA are biomarkers for assessing cognitive status in neurodegenerative diseases, such as Alzheimer’s and Huntington’s disease (80,81). The main mechanism of this effect is that mtDNA depletion induced by persistent oxidative stress leads to cognitive decline via cell apoptosis (82).
Inflammation
OSA appears to have an inflammatory component (83), but the exact mechanisms linking OSA to the inflammatory cascade are unknown. Clinically, the inflammatory component of OSA manifests as neurocognitive and behavioral deficits, with oxidative stress and inflammatory impairment, which are mediated by microglia in a model of OSA (84). Previous studies have shown that inflammation is initiated due to increased ROS production under IH cycles, as ROS induces inflammatory pathways to activate multiple proinflammatory cytokines (85).
It has also been found that IH selectively activates the proinflammatory transcription factor NF-κB, which is involved in the transcription of multiple genes in the inflammation pathway (86). According to a previous study, the transcriptional activity of NF-κB is induced by ROS in part via the alternative phosphorylation of IκBα (87). Moreover, the phosphorylation of IκBα on Tyr42 by H2O2 releases a dimer that contains p50 and RelA (88). These proteins then bind to the DNA-binding domains of NF-κB to activate NF-κB transcription (89).
However, the phosphorylation of RelA, which is influenced by ROS-dependent processes, leads to increased NF-κB activation (Fig. 1B) (90). Furthermore, patients with OSA have been reported to have increased levels of NF-κB in neutrophils and monocytes (91). These activated cells can further contribute to oxidative stress and injury via the increased release of ROS via activation of NOX2 expression (92). Therefore, these results suggested that the underlying mechanism may include a feedback cycle involving ROS and NF-κB.
When NF-κB is activated, spatial learning and memory are impaired, which may be associated with declining hippocampal long-term potentiation (LTP) and dendritic branching (93). On the other hand, inflammation decreases the efficiency of the capillary system and oxygen supply to the brain, thus reducing metabolic function and oxygen intake in neurons (94). Consequently, individuals with neuroinflammation and OSA may present with cognitive deterioration.
Apoptosis. Our previous study demonstrated that rats exposed to IH show impaired spatial learning, as well as increased apoptosis in the cortex and CA1 region of the hippocampus (69), which is associated with ER stress-induced apoptosis. While the molecular mechanisms of ROS-induced apoptosis have not been fully determined, some factors have been identified. The activity of cyclic AMP response element-binding protein (CREB), which plays a critical role in neuronal survival, has been shown to decline in the hippocampal CA1 region after exposure to IH, and is accompanied by an increase in the number of cleaved caspase-3-positive cells (95).
Moreover, increased ROS levels have been reported to reduce CREB phosphorylation and decrease the expression of brain-derived neurotrophic factor (BDNF) (96). In our previous study using a chronic IH mouse model, it was found that the expression of BDNF is significantly reduced after chronic IH treatment (70). Furthermore, BDNF plays an important role in neuronal cell apoptosis, and increased expression of BDNF ameliorates IH-induced cognitive dysfunction by decreasing neuronal apoptosis (97).
A parallel increase in the expression of HIF-1α, a key regulator of cell adaptation to hypoxia, is often observed alongside the formation of ROS (98). Under IH, the Ca2+-dependent activation of calcium-calmodulin protein kinase stimulates HIF-1α transcriptional activity and protein expression (99). Then, in turn, the stabilization of HIF-1α promotes the synthesis of ROS in mitochondria to induce cell death via the suppression of AMP-activated protein kinase (100,101).
HIF-1α stabilization, along with a decline in Bcl-2 and substantial caspase-3 expression, has been observed after IH exposure (102). Furthermore, a large number of apoptotic events have been identified in rat myocardium exposed to IH (103), and the inhibition of HIF-1α expression has been shown to decrease neuronal apoptosis (104).
Additionally, >50 oxygen sensitive genes have been identified as direct targets of HIF-1α-mediated transactivation, such as the enzymeβ-secretase 1 (BACE1) (105). It was reported that 3 days of IH treatment upregulated BACE and generated amyloid β (Aβ) via HIF-1α, thus compared with age-matched control individuals, patients with Alzheimer’s disease have a 5-fold increased risk of presenting with OSA (106). These data suggest that inhibition of HIF-1α not only suppresses apoptosis, but also reduces Aβ generation. Thus, HIF-1α may be a potential drug target for future OSA therapy.
ER stress
ER stress induced by increased ROS expression is initiated when unfolded or misfolded proteins accumulate in the ER (30). Furthermore, ER stress induces the coordinated adaptive program known as the ‘unfolded protein response’ (UPR), which involves the degradation of unfolded proteins (107). The UPR consists of three independent signaling pathways, pancreatic ER kinase signaling, activating transcription factor 6 signaling and inositol-requiring enzyme 1 signaling (108). If the accumulation of toxic unfolded and misfolded proteins results in prolonged ER stress, the perturbed and overloaded ER-folding environment persists and is associated with increased cell death (109).
Previous studies have demonstrated that ER stress is present in the brain in a mice model of OSA, accompanied by an increase in the expression of cleaved caspase-3, which is a protein marker of apoptosis (69). In addition, the expression of growth arrest and DNA damage-inducible gene 153 (GADD153), a proapoptotic protein activated by ER stress, is increased in the cortex and hippocampus of the OSA model, which is accompanied by cognitive impairment (69). Moreover, GADD153-/- mice exhibit resistance to oxidative stress (110).
In contrast, synaptic plasticity impairment is prevented by incubation with phenylbutyric acid (PBA), an inhibitor of ER stress (111). Furthermore, synapse degeneration is associated with elevatedGADD153 expression (112). Therefore, these studies suggest that ER stress also affects synaptic formation. Our previous study using a OSA model found that ER stress is involved in cognitive deficits due to LTP impairment in the hippocampus, as application of tauroursodeoxycholic acid, an inhibitor of ER stress, rescued LTP impairment by decreasing the number of apoptotic neurons and promoting the formation of synapses (69).
Disturbance of neuronal activity
Neuronal firing, especially robust persistent activity of neurons in the cortex and hippocampus, is critical in memory formation (113,114). In a previous study, it was found that hypoxia could affect membrane excitability and may involve acute modulation of ion channels, including K+-channels, Na+-channels and Ca2+-channels, which results in the depolarization or hyperpolarization of neurons (115). Moreover, hypoxia/hypoxemia underlies several pathological processes, including neuronal activity, in these regions (116).
ROS have been implicated in LTP of neural activity as they are associated with a number of K+ channels and lead to the Ca2+-dependent release of the neurotransmitter glutamate and excitotoxicity (117). ROS promote the activation of inositol 1,4,5-trisphosphate receptors in the ER, leading to Ca2+ mobilization into the cytosol, thus enhancing membrane permeability and promoting the release of glutamate (118).
Furthermore, it was observed that a higher concentration of glutamate is found in the cortex of patients with OSA (119). Thus, it is hypothesized that the release of glutamate could increase neuronal excitability, and result in neuron dysfunction and apoptosis due to excitotoxicity.
In addition, motor-evoked potentials, which are indicative of neuronal excitability, are higher in patients with OSA compared with healthy controls (120). Moreover, increased expression of c-Fos is observed in the cortex after IH treatment and is accompanied by increased apoptosis (102). Collectively, these previous studies have suggested that these neurons are under excitotoxic conditions, which leads to cognitive dysfunction, as measured by the Morris water maze (121).
Furthermore, our previous study implanted multiple electrodes into the CA1 region of the hippocampus to monitor spontaneous discharges after chronic IH treatment. It was found that the frequency of pyramidal neuron firing in the CA1 region increased after 1-2 days of IH exposure. However, this firing decreased after 14 days of IH treatment (unpublished data). These results indicated that cognitive malfunction in the OSA model may be associated with marked cellular changes over time.
reference link : https://doi.org/10.3892/etm.2020.9132
More information: G L Dunietz et al, Obstructive Sleep Apnea Treatment and Dementia Risk in Older Adults, Sleep (2021). DOI: 10.1093/sleep/zsab076



{kind=link}