










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A new gene therapy for one of the most common forms of congenital blindness was safe and improved patients’ vision, according to initial data from a clinical trial led by researchers at the Scheie Eye Institute in the Perelman School of Medicine at the University of Pennsylvania.
The therapy delivers working copies of GUCY2D to the eyes of patients who have severe vision impairments caused by mutations in the gene. Each of the first three treated patients experienced improvement in some aspects of vision, without serious side effects, according to the new study, published in the journal iScience.
“We found sustained improvements in both day and night vision, even with a relatively low dose of the gene therapy,” said study lead author Samuel G. Jacobson, MD, Ph.D., a professor of ophthalmology in the Perelman School of Medicine.
The GUCY2D gene is one of about 25 different human genes whose mutations cause problems in the retina, leading to severe vision impairment from birth or early childhood. This family of inherited retinal disorders, collectively known as Leber congenital amaurosis (LCA), accounts for a considerable portion of blindness in children worldwide.
Normal copies of GUCY2D encode an enzyme in the key pathway that light-sensitive rod and cone cells in the retina use to convert light into electrochemical signals. A lack of this enzyme blocks the recovery of this pathway, preventing the reset needed for further signaling. As a result, the signal from rod and cone cells becomes very weak – which equates to severe vision loss.
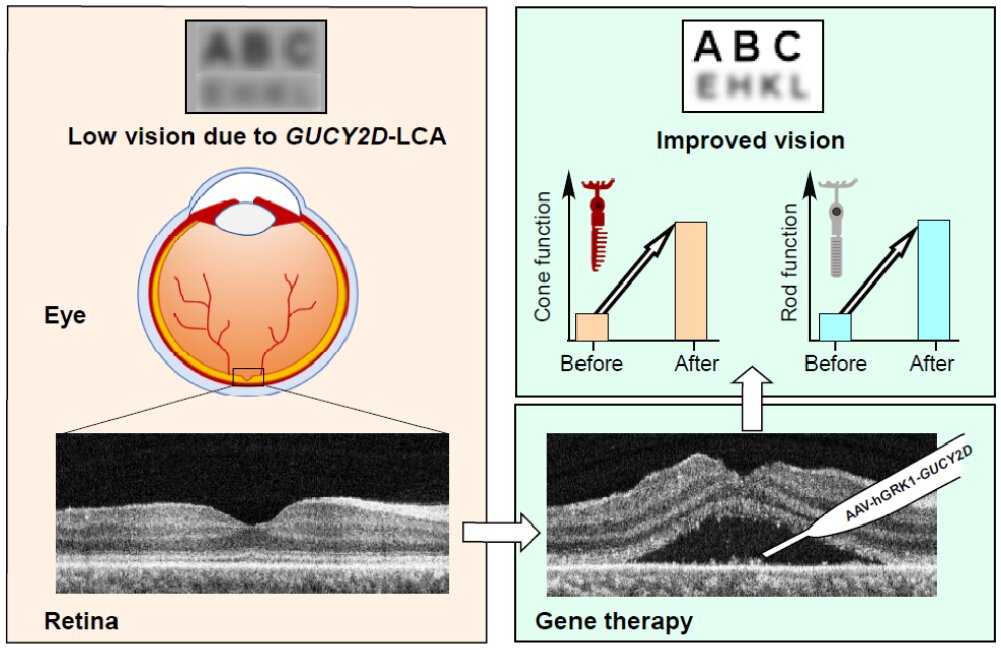
Even in adults who have lived for decades with this condition, it is often the case that many light-sensing retinal cells remain alive and intact despite their dysfunction. Thus, adding functional copies of GUCY2D via a gene therapy could get those cells working again and restore some vision.
In 2019, Jacobson and co-investigator Artur V. Cideciyan, Ph.D., a research professor of ophthalmology in the Perelman School of Medicine, began the first clinical trial of a GUCY2D gene therapy, a solution of a harmless virus that carries the gene and is injected beneath the retina – initially in just one eye per patient.
They are following each patient for two years after treatment. In the new report, they described their findings after nine months in the first three patients treated.
The first patient experienced a substantial increase in light-sensitivity in rod cells, which are more light-sensitive than cone cells and are chiefly responsible for low-light or “night vision.” This patient also showed improved pupil responses to light.
The second patient showed a smaller but sustained increase in light-sensitivity in rod cells, starting about two months after the gene therapy.
The third patient showed no improvement in rod cell sensitivity, but did show significantly improved visual acuity over the nine-month follow-up period, an improvement that the researchers tied to better function in the patient’s cone cells, the predominant cells for daylight and color vision.
“These initial results from the first-ever trial of a GUCY2D gene therapy are very encouraging and will inform our ongoing and future trials of this therapy,” said Cideciyan.
There were no serious adverse side effects, and any side effects that occurred in the patients’ retinas resolved.
The gene therapy dose used in these first three patients was the lowest of the doses the researchers plan to use in the study, so they are hoping to see continued safety and greater efficacy in later-enrolled patients who will receive higher doses.
The ongoing clinical trial is registered at clinicaltrials.gov as trial NCT03920007.
Jacobson and Cideciyan in earlier studies have reported success for gene-based therapies against two other forms of Leber congenital amaurosis involving other genes.
Inherited retinal disorder (IRD) is a leading cause of blindness,1 and includes disorders such as retinitis pigmentosa (RP), cone/cone-rod dystrophy (CORD), macular dystrophy (MD), Stargardt disease (STGD), Leber congenital amaurosis (LCA) and others.1–6 IRD is characterized by heterogeneity both in the clinical and genetic aspects, with different inheritance patterns, including autosomal dominant (AD), autosomal recessive (AR), X-linked, and mitochondrial inheritance.7–9
Significant clinical and genetic overlap is well-known in the spectrum of IRD, and diverse clinical phenotypes, including CORD, MD, STGD, RP, and LCA, can manifest as a result of pathogenic variants in a single gene (e.g., ABCA4, BEST1, PRPH2, RPGR, CRX, GUCY2D, RS1, POC1B, PROM1, CNGA3, CNGB3).2,3,7,8,10–18
GUCY2D, denoted as guanylate cyclase 2D (OMIM: 600179), is located on 17p13.1 and contains 20 exons and encodes one of the two retinal membrane guanylyl cyclase isozymes expressed in photoreceptors.19,20 Retinal membrane guanylyl cyclase isozymes synthesize the intracellular messenger of photoreceptor excitation, cyclic guanosine monophosphate, which is regulated by the intracellular Ca2+-sensor proteins of guanylate cyclase-activating proteins.19–26
RetGCs and guanylate cyclase-activating proteins are responsible for the Ca2+ -sensitive restoration of cyclic guanosine monophosphate levels after the light activation of the phototransduction cascade.26
A locus and gene for LCA was first mapped and identified as GUCY2D (LCA1) in 1995 and 1996.19,27 Since then, more than 200 variants in the GUCY2D gene have been associated with a wide range of different phenotypes of IRDs.9,19,20,28–39 Sharon et al. reported that 88% of GUCY2D-associated retinal disorder (GUCY2D-RD) is AR-LCA, whereas pathogenic heterozygous missense GUCY2D variants cause AD-CORD.20
In that, pathogenic GUCY2D variants are one of the major causes of LCA, as well as a major cause of AD-CORD.20 Recently, Stunkel et al. identified five patients with AR congenital night blindness caused by biallelic GUCY2D variants, which may slowly progress to mild retinitis pigmentosa.40 Thus, AR-LCA, AD-CORD, and AR congenital night blindness are the main clinical presentations of GUCY2D-RD.
Studies of GUCY2D-RD have been conducted separately for each phenotype, such as CORD or RP/LCA; thus, it has been hard to comprehensively understand the disorder with diverse clinical manifestations and different modes of inheritance. To grasp the whole picture of GUCY2D-RD, large cohort studies with standardized clinical and genetic investigations for IRD in total are required.
The purpose of this study was to characterize the clinical and molecular genetic features of GUCY2D-RD in a large nationwide cohort of Japanese subjects diagnosed with IRD.
reference link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7408927/
More information: Samuel G. Jacobson et al. Safety and Improved Efficacy Signals following Gene Therapy in Childhood Blindness Caused by GUCY2D Mutations, iScience (2021). DOI: 10.1016/j.isci.2021.102409



{kind=link}