")










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Gli scienziati sanno da tempo che le distintive proteine ”spike” di SARS-CoV-2 aiutano il virus a infettare il suo ospite attaccandosi alle cellule sane. Ora, un nuovo importante studio mostra che svolgono anche un ruolo chiave nella malattia stessa.
Il documento, pubblicato il 30 aprile 2021, su Circulation Research, mostra anche in modo conclusivo che COVID-19 è una malattia vascolare , dimostrando esattamente come il virus SARS-CoV-2 danneggia e attacca il sistema vascolare a livello cellulare.
I risultati aiutano a spiegare l’ampia varietà di complicazioni apparentemente non collegate di COVID-19 e potrebbero aprire la porta a nuove ricerche su terapie più efficaci.
“Molte persone lo considerano un disturbo respiratorio
malessere, ma è davvero una malattia vascolare “, afferma il professore assistente di ricerca Uri Manor, che è co-autore senior dello studio. “Questo potrebbe spiegare perché alcune persone hanno ictus e perché alcune persone hanno problemi in altre parti del corpo. La cosa in comune tra loro è che hanno tutte basi vascolari “.
I ricercatori di Salk hanno collaborato con scienziati dell’Università della California a San Diego sull’articolo, tra cui il co-primo autore Jiao Zhang e il co-autore senior John Shyy, tra gli altri.
Sebbene i risultati stessi non siano del tutto una sorpresa, il documento fornisce una chiara conferma e una spiegazione dettagliata del meccanismo attraverso il quale la proteina danneggia le cellule vascolari per la prima volta.
C’è stato un crescente consenso sul fatto che SARS-CoV-2 influenzi il sistema vascolare , ma non è stato compreso esattamente come abbia fatto. Allo stesso modo, gli scienziati che studiano altri coronavirus sospettano da tempo che la proteina spike abbia contribuito a danneggiare le cellule endoteliali vascolari, ma questa è la prima volta che il processo è stato documentato.
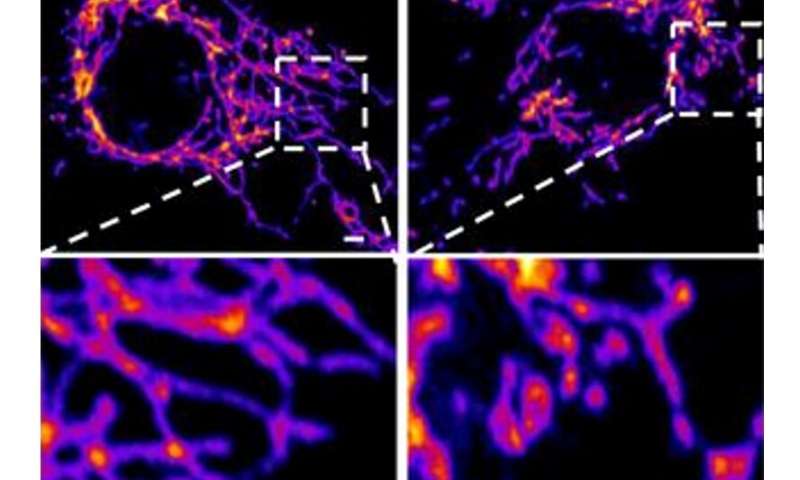
Nel nuovo studio, i ricercatori hanno creato uno “pseudovirus” circondato dalla classica corona di proteine spike SARS-CoV-2 , ma che non conteneva alcun virus effettivo. L’esposizione a questo pseudovirus ha provocato danni ai polmoni e alle arterie di un modello animale, dimostrando che la sola proteina spike era sufficiente a causare la malattia. I campioni di tessuto hanno mostrato infiammazione nelle cellule endoteliali che rivestono le pareti delle arterie polmonari.
Il team ha quindi replicato questo processo in laboratorio, esponendo cellule endoteliali sane (che rivestono le arterie) alla proteina spike. Hanno dimostrato che la proteina spike danneggiava le cellule legando ACE2.
Questo legame ha interrotto la segnalazione molecolare di ACE2 ai mitocondri (organelli che generano energia per le cellule), provocando il danneggiamento e la frammentazione dei mitocondri.
Studi precedenti hanno mostrato un effetto simile quando le cellule sono state esposte al virus SARS-CoV-2, ma questo è il primo studio a dimostrare che il danno si verifica quando le cellule sono esposte alla proteina spike da sole.
“Se rimuovi le capacità di replicazione del virus, ha ancora un effetto dannoso importante sulle cellule vascolari, semplicemente in virtù della sua capacità di legarsi a questo recettore ACE2, il recettore della proteina S, ora famoso grazie a COVID”, spiega Manor . “Ulteriori studi con proteine mutanti spike forniranno anche nuove informazioni sull’infettività e la gravità dei virus mutanti SARS CoV-2”.
Con l’avanzare dell’età, la velocità e la forza della loro risposta immunitaria si indeboliscono a causa della perdita di alcuni tessuti immunitari come il timo, nonché del metabolismo energetico più povero a livello cellulare con i mitocondri. L’energia nella cellula si presenta sotto forma di adenosina trifosfato (ATP) ed è prodotta dai mitocondri quando la cellula è alimentata con l’ossigeno, così come dalla glicolisi in assenza di ossigeno.
È anche noto che i mitocondri interagiscono con le particelle virali quando infettano cellule ospiti umane, coinvolgendo il rilascio di interferone e citochine, stimolando l’infiammazione e influenzando la sopravvivenza e la replicazione virale (Khan et al., 2015; Tiku et al., 2020). Lo studio dell’immunità mitocondriale contro il SARS-CoV-2 può fornire informazioni sul motivo per cui gli individui più anziani, con una ridotta efficienza mitocondriale, forse peggio equipaggiati per affrontare COVID-19.
Lo scopo del nostro studio è esplorare il legame molecolare tra i mitocondri negli individui anziani e SARS-CoV-2. Inoltre, abbiamo anche evidenziato il ruolo di varie comorbidità legate all’età come diabete, obesità e malattie neurologiche nell’aumento dei tassi di mortalità tra gli anziani con COVID-19. Esploriamo anche i trattamenti attuali, gli stili di vita e le misure di sicurezza che possono aiutare a proteggere contro COVID-19.
Mitocondri e immunità
I mitocondri sono organelli con una doppia membrana che funge da fonte primaria di produzione di energia di una cellula sotto forma di ATP e contribuisce all’omeostasi, alla proliferazione cellulare, alla morte cellulare e alla sintesi di aminoacidi, lipidi e nucleotidi. In caso di infezione, i mitocondri contribuiscono all’immunità coinvolgendo il sistema dell’interferone, alterandone la struttura e inducendo la morte cellulare programmata (apoptosi; Ohta e Nishiyama, 2011).
Segnalazione dell’interferone e mitocondri
In caso di infezione virale, il sistema immunitario innato dell’ospite riconosce determinati schemi, come sequenze di acidi nucleici virali o proteine virali, quando si attaccano ai recettori sulle membrane cellulari dell’ospite, a livello intracellulare ed extracellulare. Il loro riconoscimento attiva le vie di segnalazione che portano alla risposta infiammatoria. Esistono diversi tipi di recettori a cui si attaccano questi componenti virali, inclusi i recettori toll-like e i recettori gene-I-I inducibili dall’acido retinoico (RLRs; Takeuchi e Akira, 2009).
I recettori Toll-like sono coinvolti nell’attivazione dell’interferone di tipo I, una citochina infiammatoria e nella produzione di chemochine e si trovano sulla superficie cellulare, sull’endosoma e sulle membrane del reticolo endoplasmatico (Takeuchi e Akira, 2009). Gli RLR sono i recettori citosolici che avviano la produzione di interferone di tipo I nelle cellule non immunitarie (Takeuchi e Akira, 2009) e possono essere trovati sui mitocondri (Tal e Iwasaki, 2009).
Questi RLR rilevano l’RNA virale nel citoplasma e sono coinvolti nel riconoscimento di virus a RNA come i paramixovirus, il virus dell’encefalite giapponese, il virus dell’influenza e i picornavirus (Kato et al., 2006). Tuttavia, alcuni sottotipi di RLR possono essere coinvolti anche nel rilevamento di alcuni virus a DNA.
Per esempio, l’adenovirus e il virus Herpes Simplex di tipo 1 hanno una RNA polimerasi III dipendente dal DNA che influenza gli RLR e stimola la produzione di interferone-β, e il virus Epstein-Barr produce piccoli frammenti di RNA che possono attivare RLR (Samanta et al., 2006; Cheng et al., 2007; Chiu et al., 2009).
Un componente della via di segnalazione derivante dall’attivazione della RLR è una molecola nota come proteina di segnalazione antivirale mitocondriale (MAVS; Kawai et al., 2005). MAVS si trova sulla membrana mitocondriale esterna (OMM; Seth et al., 2005) e, dopo l’attivazione, innesca fattori di trascrizione che si tradurranno in una produzione aggiuntiva di interferone (Zhang et al., 2014).
Oltre al MAVS, la proteina associata ai mitocondri chiamata “stimolatore dei geni dell’interferone” e la proteina mitocondriale mitofusina 2 sono anche coinvolte nelle cascate RLR o lavorano con MAVS (Ishikawa e Barber, 2008; Yasukawa et al., 2009).
Questa prova dimostra che i mitocondri sono una parte importante della segnalazione dell’interferone nel sistema immunitario. Alcuni virus alterano i livelli di MAVS per prevenire la produzione di interferone; il virus dell’influenza A (Varga et al., 2012), il virus del morbillo (Xia et al.,
Fissione mitocondriale e modulazione della fusione
I virus possono manipolare la fissione e la fusione mitocondriale per favorire la sopravvivenza virale (Holder e Reddy, 2020). I mitocondri possono alterare la loro struttura attraverso la fissione e la fusione del loro OMM e della membrana mitocondriale interna (IMM), mediante funzioni che coinvolgono le GTPasi correlate alla dinamina (Tiku et al., 2020).
La fusione dell’OMM è mediata dalle proteine Mitofusina 1 e Mitofusina 2 tramite idrolisi GTP e la fusione dell’IMM è mediata dall’atrofia ottica proteica 1, che è una GTPasi presente nell’IMM (Tiku et al., 2020). La fusione mitocondriale è necessaria per lo scambio di DNA, proteine e metaboliti mitocondriali (Archer, 2013).
D’altra parte, la fissione dell’OMM è mediata dalla proteina citosolica GTPasi dinamina 1 (Drp1) tramite idrolisi GTP (Mears et al., 2011). Dopo aver trovato un sito di scissione mitocondriale, Drp1 interagisce con il fattore di fissione mitocondriale e le proteine della dinamica mitocondriale 49 e 51 per restringere e tagliare l’OMM (Mears et al., 2011; Loson et al., 2013). I meccanismi della fissione mitocondriale non sono ben compresi.
La fissione è necessaria per rimuovere le parti danneggiate dei mitocondri da eliminare dalla mitofagia (autofagia dei mitocondri) ed è necessaria durante la replicazione del ciclo cellulare (Mao e Klionsky, 2013). Pertanto, la fissione potenziata di solito porta ad un aumento della mitofagia.
Alcuni virus possono promuovere la fusione mitocondriale per ridurre la risposta pro-infiammatoria dell’interferone contro i virus attraverso un meccanismo che coinvolge l’inibizione della MAVS da parte della mitofusina 2. Ad esempio, il virus della dengue stimola la fusione mitocondriale tramite la sua proteina non strutturale NS4B (Barbier et al., 2017) e l’HIV migliora la fusione tramite la sua proteina busta gp120 (Fields et al., 2016).
Il coronavirus SARS (SARS-CoV-1) migliora la fusione tramite il suo fattore di virulenza ORF-9b (Shi et al., 2014). Questi fattori di virulenza riducono i livelli di Drp1, la proteina che induce la fissione, portando così a una fusione mitocondriale sbilanciata, che è guidata dalla mitofusina 2.
Poiché la mitofusina 2 interagisce con e inibisce MAVS, che tipicamente aumenta gli interferoni (Yasukawa et al., 2009) questo può ostacolare la risposta dell’interferone. È interessante notare che SARS-CoV-1 utilizza lo stesso ORF-9b anche per ridurre direttamente i livelli di MAVS, il che riduce ulteriormente la risposta dell’interferone (Shi et al., 2014).
Alcuni virus inducono la fissione mitocondriale per aumentare la mitofagia e alterare il tasso di apoptosi, di solito tramite up-regolazione o attivazione di Drp1 e / o degradazione o inibizione di MAVS (Khan et al., 2015). Tra questi vi è il virus dell’epatite C tramite le sue proteine e le proteine E1-E2 (Kim et al., 2014), il citomegalovirus umano tramite la proteina virale vMIA (McCormick et al., 2003) e il virus dell’epatite B tramite la proteina virale HBx (Kim et al., 2013). La fissione mediata da questi tre particolari virus porta all’inibizione dell’apoptosi in modo che il virus possa sopravvivere più a lungo e replicarsi ulteriormente (McCormick et al., 2003; Kim et al., 2013, 2014).
A causa della modulazione da parte dei virus sulla fusione e fissione mitocondriale, la loro presenza può portare a livelli di energia alterati attraverso il conteggio e la forma mitocondriale. I virus che causano la fissione mitocondriale e portano all’inibizione dell’apoptosi possono consentire alle particelle virali di sopravvivere incolume più a lungo. Molti pazienti spesso si sentono deboli per mancanza di energia quando sono infettati da una malattia virale. Ciò può essere dovuto alla più scarsa produzione di energia mitocondriale come risultato dell’aumentata fissione.
Morte cellulare
L’apoptosi, o morte cellulare programmata, è un’altra importante funzione della cellula influenzata dai mitocondri. Esiste una via estrinseca per attivare l’apoptosi, controllata da alcuni ligandi che si legano ai recettori della “morte”, e una via intrinseca controllata dai mitocondri (Brenner e Mak, 2009).
In questo percorso intrinseco, la membrana mitocondriale è permeata e il potenziale di membrana mitocondriale (MMP) viene interrotto quando le proteine dello spazio intermembrana si riversano nel citoplasma (Shawgo et al., 2008). Queste proteine includono il citocromo c (Liu et al., 1996), caspase-9 (Du et al., 2000) e il fattore 1 di attivazione della proteasi dell’apoptosi che lavorano insieme per formare un apoptosoma, che stimola le caspasi finali a portare a termine la morte cellulare procedura (Cain et al., 2002).
Si ritiene che la destabilizzazione della MMP, che porta all’apoptosi, avvenga tramite vari meccanismi. In primo luogo, c’è il meccanismo della permeabilizzazione selettiva dell’OMM (Kuwana et al., 2002). Bax e Bak, proteine che costituiscono la sottofamiglia proapoptotica Bax delle proteine Bcl-2, servono come pori sulla membrana mitocondriale per mantenere la MMP e rilasciano citocromo ce calcio dall’interno dei mitocondri (Nutt et al., 2002).
Quando un’altra sottofamiglia proapoptotica nota come sottofamiglia solo BH3 si attacca a Bax / Bak attivato, aumentano la permeabilità e aumentano la possibilità di apoptosi (Chen et al., 2005). D’altra parte, la sottofamiglia antiapoptotica Bcl-2 (inclusi Bcl-2 e Bcl-xL) può legarsi alle proteine Bax / Bak attivate e inibire Bax / Bak formando un complesso antiapoptotico e portando a una diminuzione dell’apoptosi (Shawgo et al., 2008).
Un secondo meccanismo coinvolge la composizione del doppio strato lipidico della membrana mitocondriale e coinvolge Bax che crea una rapida riorganizzazione dei lipidi che porta allo stress strutturale e alla formazione di buchi (Terrones et al., 2004). L’ultimo meccanismo coinvolge la stimolazione del complesso dei pori di transizione di permeabilità (PTPC) nell’IMM (Shimizu et al., 1999). Ciò è innescato da una sovrabbondanza di specie di calcio o ossigeno reattivo (Deniaud et al., 2008) e può essere influenzato dalle proteine di tutte le parti dei mitocondri.
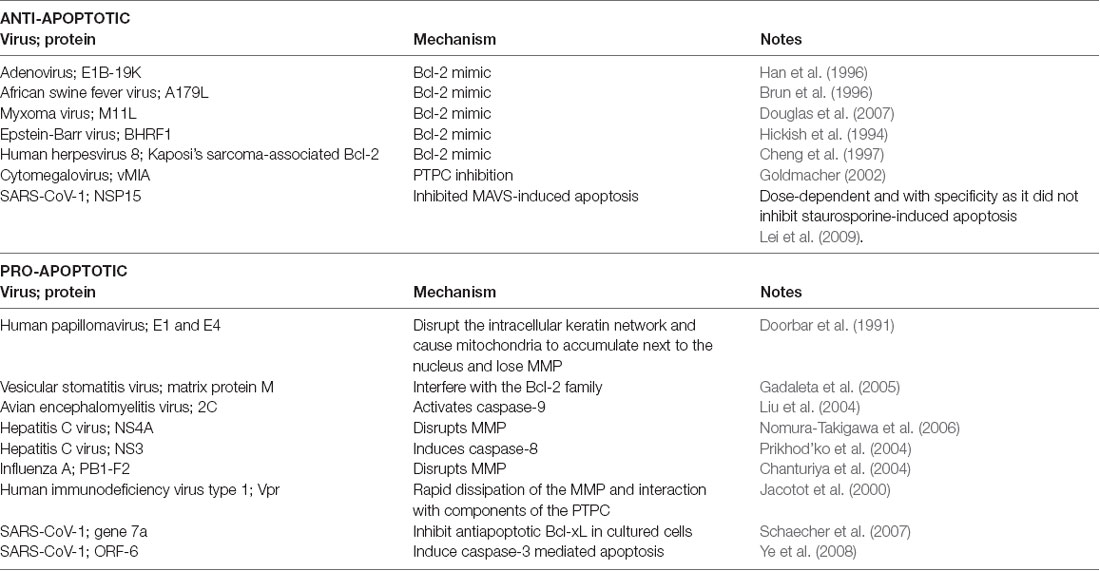
I virus possono utilizzare proteine virali che imitano i membri della famiglia Bcl-2 e altri fattori coinvolti nelle vie di apoptosi per manipolare la durata della vita della cellula come meglio credono. La tabella 1 mostra alcuni esempi comuni per illustrare questo punto.
SARS-CoV-2 e mitocondri
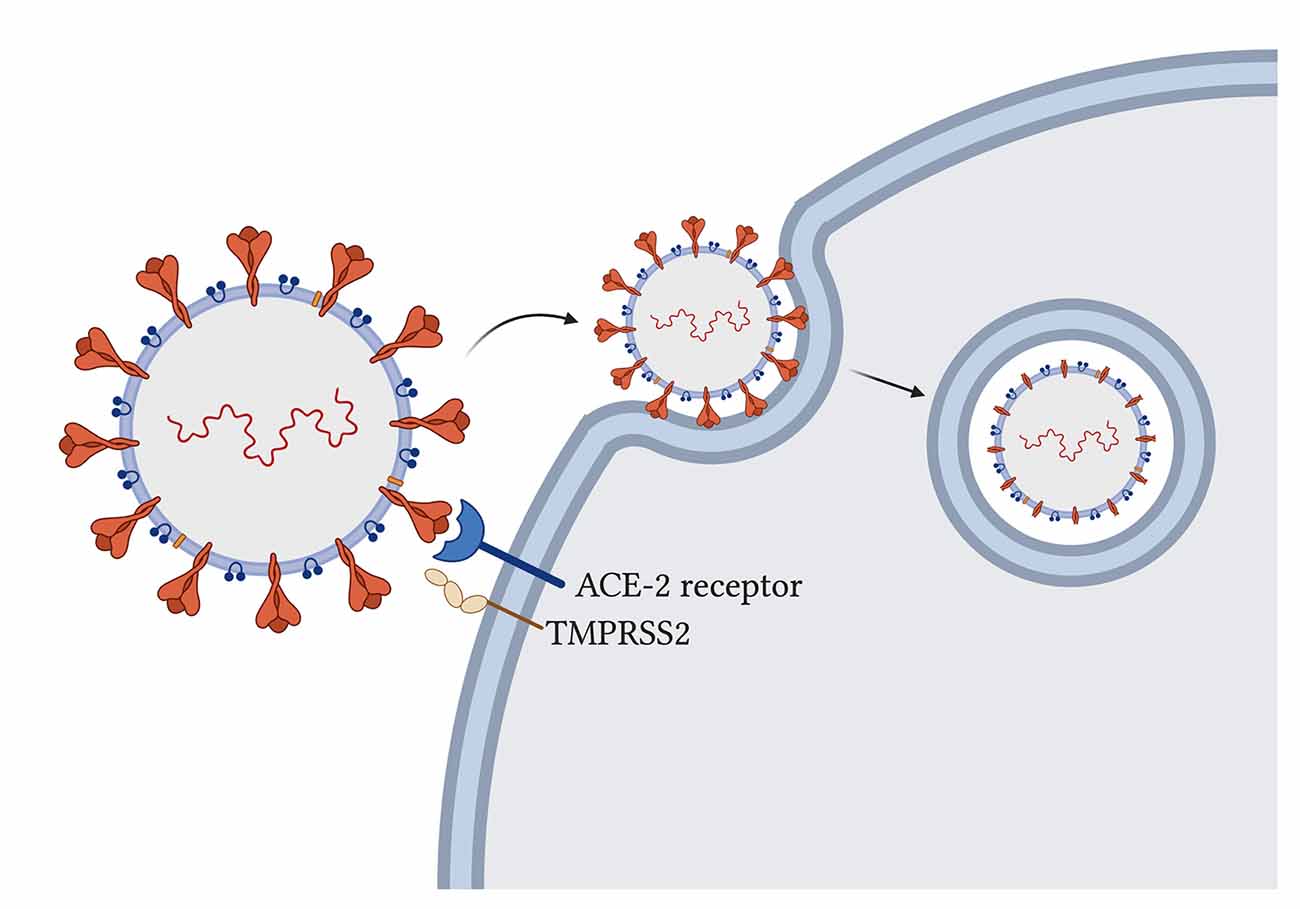
Il nuovo SARS-CoV-2 utilizza la sua glicoproteina spike sul recettore ospite dell’enzima di conversione dell’angiotensina-2 (ACE-2) (Cao et al., 2020) per entrare nelle cellule ospiti umane e la serina proteasi transmembrana ospite 2 (TMPRSS2) per innescare la proteina spike per l’attacco (Hoffmann et al., 2020; Figura 2).
La particella virale entra nella cellula tramite endocitosi ed è stato proposto che la proteina spike debba essere scissa dagli enzimi ospiti affinché avvenga l’ingresso virale (Ou et al., 2020). L’ACE-2 influenza le funzioni mitocondriali e una mancanza di ACE-2 è correlata alla diminuzione della produzione di ATP e all’alterata attivazione della NADPH ossidasi 4 nei mitocondri, che viene normalmente utilizzata per la produzione di ROS (Singh et al., 2020) che possono entrambi proteggere la cellula distruggendo gli agenti patogeni o inducendo la cellula infetta ad andare in apoptosi.
Con il virus SARS-CoV-2 che utilizza i recettori ACE-2 per il suo ingresso, la disponibilità di ACE-2 per le sue normali funzioni può essere ridotta e contribuire allo sviluppo dei sintomi.
Inoltre, alcuni studi hanno suggerito che il TMPRSS2 da SARS-CoV-2 influenza anche la funzione mitocondriale agendo sul recettore alfa correlato agli estrogeni, che è un recettore nucleare che regola la trascrizione delle funzioni mitocondriali e l’omeostasi energetica (Xu et al., 2018 ; Hoffmann et al., 2020; Singh et al., 2020).
Una volta all’interno della cellula, SARS-CoV-2 innesca una massiccia risposta infiammatoria. Attraverso le funzioni immunitarie innate innescate dall’infezione virale sopra descritte, citochine come TNF-α, INF-γ e interleuchina-10 arrivano alle cellule infette e causano un aumento della produzione di ROS mitocondriali attraverso la sovraregolazione dell’espressione genica e la modulazione della catena di trasporto degli elettroni (Saleh et al., 2020).
Il ROS mitocondriale stimola quindi la produzione di citochine proinfiammatorie aggiuntive (Li et al., 2013) mentre il virus continua a persistere, portando infine a una “tempesta di citochine” in cui l’eccessiva infiammazione può causare danni fatali se l’immunità adattativa non prende il sopravvento in tempo.
La risposta immunitaria induce anche i mitocondri a deviare parte dell’energia dalla produzione di ATP per contribuire alla produzione di ROS, che può danneggiare i mitocondri in quantità schiaccianti, portando alla permeabilizzazione della membrana e all’apoptosi (Saleh et al., 2020). Se i mitocondri gravemente danneggiati rilasciano il loro contenuto nello spazio citosolico, stimolano la produzione di più citochine come IL-1β e IL-6 che sono i tratti distintivi di COVID-19 (Saleh et al., 2020).
Un altro meccanismo di distruzione mitocondriale impiegato da SARS-CoV-2 coinvolge la ferritina, come evidenziato dagli alti livelli di ferritina in quelli con esiti gravi (Aguirre e Culotta, 2012). Un mitocondrio normalmente funzionante utilizza questo ferro per produrre eme, creare ammassi di ferro-zolfo e immagazzinare come ferritina mitocondriale (Saleh et al., 2020), ma un sovraccarico di ferro può portare a stress ossidativo e compromettere la funzione mitocondriale riducendo il consumo di ossigeno di i mitocondri (Tang et al., 2020). Inoltre, il sovraccarico di ferritina può interrompere la tolleranza al glucosio in queste cellule con stress ossidativo mitocondriale (Tang et al., 2020), che ha implicazioni per i pazienti diabetici.
È stato teorizzato che SARS-CoV-2 utilizzi vescicole a doppia membrana derivate da membrane mitocondriali per nascondersi e proteggersi all’interno della cellula (Singh et al., 2020). Questa teoria si basa sull’evidenza dell’HIV utilizzando vescicole a doppia membrana derivate da ER (Somasundaran et al., 1994) e un’osservazione che una mutazione puntiforme nel coronavirus nei roditori ha dimostrato di diminuire le vescicole derivate da ER e aumentare la localizzazione del virus ai mitocondri allo stesso tempo (Clementz et al., 2008).
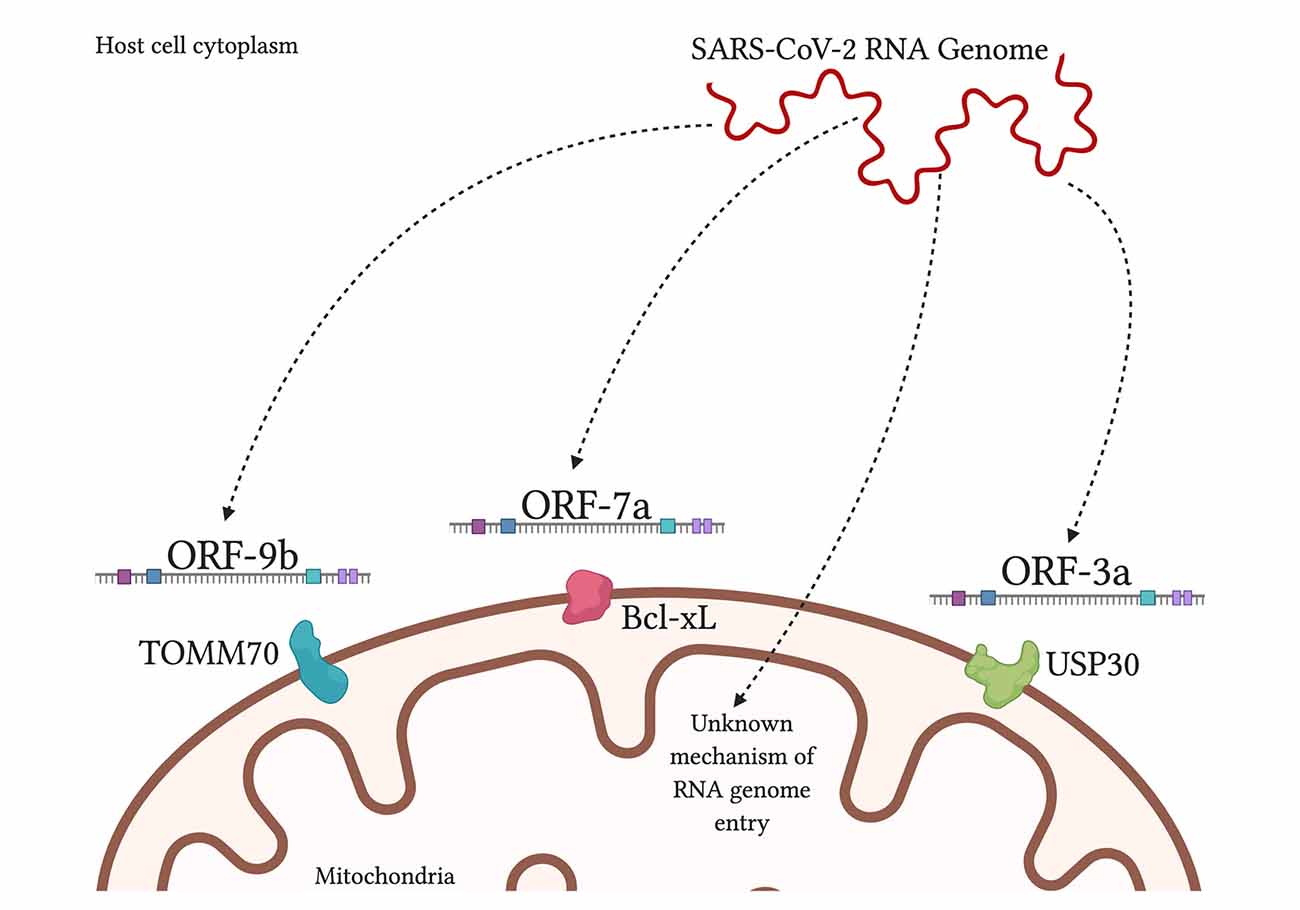
Inoltre, uno studio ha trovato regioni 5 ′ e 3 ′ non tradotte su SARS-CoV-2 uniche per la localizzazione mitocondriale, sebbene sia necessario lavorare ulteriormente su questa scoperta (Wu et al., 2020). Quando si confrontano SARS-CoV-1 e SARS-CoV-2, entrambi contengono frame di lettura aperti ORF-9b, ORF-7a e ORF-8b, che si localizzano ai mitocondri, nel caso di SARS-CoV-1 , per alterare la funzione MAVS e la funzione mitocondriale (Chen et al., 2007; Shi et al., 2014).
SARS-CoV-2 aveva inoltre ORF-3a presente ma ORF-3b assente (Singh et al., 2020; Figura 3). Incoraggiando la formazione di vescicole a doppia membrana dalla membrana mitocondriale o anche dalla membrana ER, SARS-CoV-2 può evitare in sicurezza gli attacchi dei ROS e delle proteasi dell’ospite che ne minacciano la sopravvivenza. Nel frattempo, il ROS si attarda e può attaccare i tessuti sani.
Esistono prove del fatto che SARS-CoV-1 ORF-9b causi la fusione mitocondriale dalla degradazione di Drp1 da parte dei proteasomi (Shi et al., 2014; Holder e Reddy, 2020), e date le somiglianze nel genoma, è probabile che anche il SARS-CoV-2 ORF-9b sta riducendo la quantità di Drp1, portando a una maggiore fusione. La fusione mitocondriale, che in parte avviene tramite mitofusina 2, può portare a una risposta interferone ostacolata tramite l’inibizione di MAVS (Yasukawa et al., 2009).
Sebbene ciò suggerisca che i numeri ridotti di interferone possano eliminare specificamente l’apoptosi indotta dall’interferone (Chawla-Sarkar et al., 2003), dobbiamo considerare che SARS-CoV-1 è noto per indurre l’apoptosi attraverso altri fattori come ORF-6 e -7a (Schaecher et al., 2007; Ye et al., 2008). Il confronto di entrambi i virus della SARS indica che SARS-CoV-2 può indurre l’apoptosi quando il suo fabbisogno per la cellula ospite umana è terminato.
Inoltre, ci sono alcune prove di ferroptosi o apoptosi indotta da ferritina con sovraccarico di ferro. I mitocondri difettosi non possono metabolizzare il ferro come farebbero normalmente, portando ad accumulo di ferro e ferroptosi (Saleh et al., 2020). Tutto ciò implica un maggior numero di morti cellulari con COVID-19.
SARS-CoV-2 può anche interferire con la conta piastrinica e la coagulazione, in particolare con l’aumento della coagulabilità e la diminuzione della conta piastrinica all’aumentare della gravità del COVID-19 (Tang et al., 2020; Terpos et al., 2020). Oltre all’aumento del rischio di ictus, l’aumento della coagulazione e la diminuzione delle piastrine stanno compromettendo la capacità delle cellule di subire la mitofagia (Lee et al., 2016).
Quando le piastrine non possono subire la mitofagia, subiscono l’apoptosi, che porta ad una maggiore formazione di trombi; questo è particolarmente vero nei pazienti diabetici che soffrono di stress ossidativo che distrugge i loro mitocondri ma ostacola la mitofagia (Lee et al., 2016). I pazienti COVID-19 soffrono di iperinfiammazione e accumulo di ferro, entrambi stressanti per le piastrine e quindi contribuiscono alla diminuzione della conta piastrinica (Saleh et al., 2020).
Gli uomini hanno avuto esiti più gravi con COVID-19 rispetto alle donne. Sebbene la causa sia sconosciuta, è stato ipotizzato che sia coinvolto il recettore TMPRSS2 (Singh et al., 2020). TMPRSS2 può essere indotto dagli androgeni, ma non dagli estrogeni, e localizzarsi nei mitocondri per regolare la funzione mitocondriale (Singh et al., 2020). Anche gli individui più anziani hanno avuto esiti peggiori. L’invecchiamento è accompagnato da una diminuzione della funzione mitocondriale, che ha dimostrato di peggiorare la gravità della malattia virale ed è anche collegata a numerose malattie legate all’età.
link di riferimento: https://www.frontiersin.org/articles/10.3389/fnagi.2020.614650/full
Ulteriori informazioni: Yuyang Lei et al, SARS-CoV-2 Spike Protein altera la funzione endoteliale tramite la downregulation di ACE 2, Circulation Research (2021). DOI: 10.1161 / CIRCRESAHA.121.318902



{kind=link}