










 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



On July 27, 2021, CDC released updated guidance on the need for urgently increasing COVID-19 vaccination coverage and a recommendation for everyone in areas of substantial or high transmission to wear a mask in public indoor places, even if they are fully vaccinated. CDC issued this new guidance due to several concerning developments and newly emerging data signals.
- In late June, our 7-day moving average of reported cases was around 12,000. On July 27, the 7-day moving average of cases reached over 60,000. This case rate looked more like the rate of cases we had seen before the vaccine was widely available.
Second, new data began to emerge that the Delta variant was more infectious and was leading to increased transmissibility when compared to other variants, even in vaccinated individuals. This includes recently published data from CDC and our public health partners, unpublished surveillance data that will be publicly available in the coming weeks, information included in CDC’s updated Science Brief on COVID-19 Vaccines and Vaccination, and ongoing outbreak investigations linked to the Delta variant.
Delta is currently the predominant strain of the virus in the United States. Below is a high-level summary of what CDC scientists have recently learned about the Delta variant. More information will be made available when more data are published or released in other formats.
Infections and Spread
The Delta variant causes more infections and spreads faster than early forms of SARS-CoV-2
- The Delta variant is more contagious: The Delta variant is highly contagious, more than 2x as contagious as previous variants.
- Some data suggest the Delta variant might cause more severe illness than previous strains in unvaccinated persons. In two different studies from Canada and Scotland, patients infected with the Delta variant were more likely to be hospitalized than patients infected with Alpha or the original virus strains.
- Unvaccinated people remain the greatest concern: Although breakthrough infections happen much less often than infections in unvaccinated people, individuals infected with the Delta variant, including fully vaccinated people with symptomatic breakthrough infections, can transmit it to others. CDC is continuing to assess data on whether fully vaccinated people with asymptomatic breakthrough infections can transmit. However, the greatest risk of transmission is among unvaccinated people who are much more likely to contract, and therefore transmit the virus.
- Fully vaccinated people with Delta variant breakthrough infections can spread the virus to others. However, vaccinated people appear to be infectious for a shorter period: Previous variants typically produced less virus in the body of infected fully vaccinated people (breakthrough infections) than in unvaccinated people. In contrast, the Delta variant seems to produce the same high amount of virus in both unvaccinated and fully vaccinated people. However, like other variants, the amount of virus produced by Delta breakthrough infections in fully vaccinated people also goes down faster than infections in unvaccinated people. This means fully vaccinated people are likely infectious for less time than unvaccinated people.

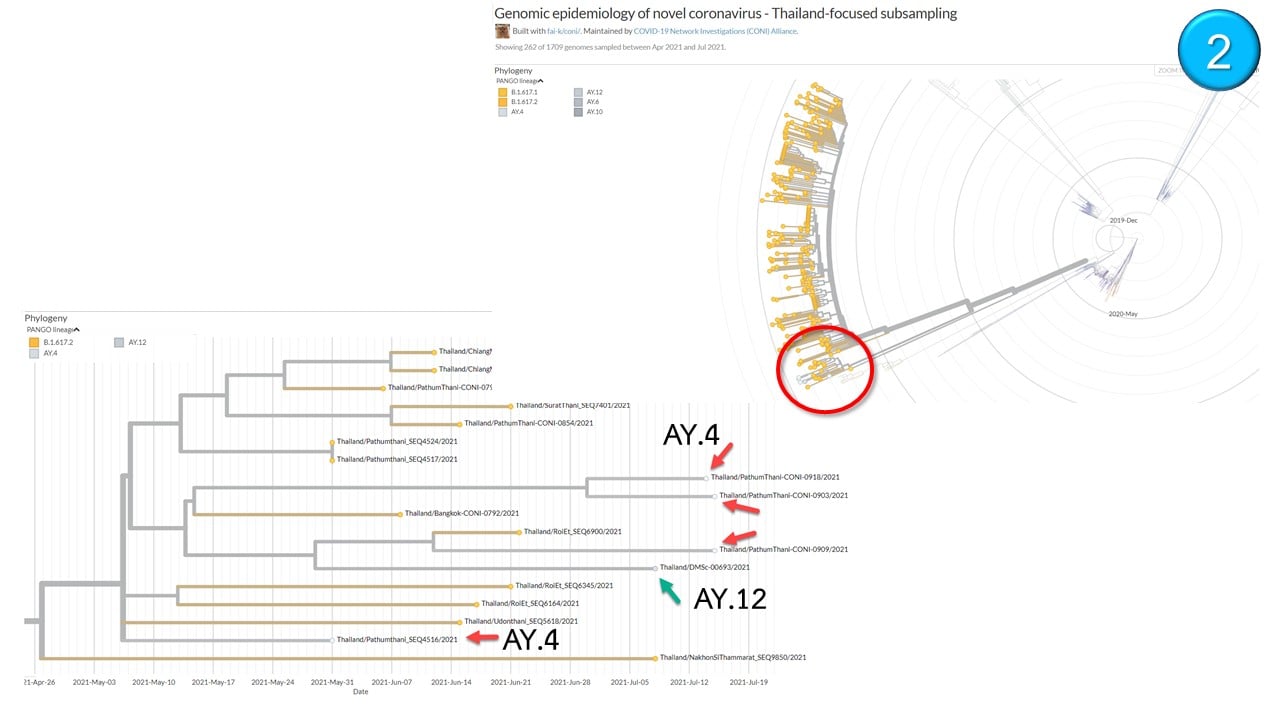
Four Delta Second Generation Variants Found In Thailand ie AY.4, AY.6, AY.10 and AY.12
Thailand’s Center For Medical Genomics has announced that 4 different second generation Delta variants bearing unique mutations have been identified in Thailand so far in 14 different individuals.
According to local Thai experts, the Delta AY.4 variant was found in a total of 9 gene sequencing cases in Thailand with Pathumthani province reporting 4 cases and one each case in the following provinces-Buriram, Kamphaeng Phet, Chiang Mai, Samut Prakan and Chonburi.
In Bangkok, one case each of the AY.6, AY.10 and AY.12 Delta variants was detected while the province of Surat Thani reported 2 cases of the AY.12 variant.
Credit: Thailand Center For Medical Genomics
To date globally, a total of 27 different second and third generation Delta variants have been identified in the last 96 hours. 22 of these are named as Delta sub-species AY.1 to 22 while the rest are still being studied. In the naming nomenclature the Delta variant (B.1.617.2) sub-species AY.1 would be labeled as B.1.617.2.1
Global researchers are worried as some of these variants are possessing concerning mutations on them and more detailed studies are underway to understand how these mutations could cause differences in pathogenesis patterns and as well as enhancing viral fitness, virulence and also transmissibility.
Local Thai authorities also reported that the current predominance of the various VOCs or variants of concern in the country are as follows:
B.1.1.7 (alpha) 11%
B.1.351 (Beta) 14%
B.1.617.2 (Delta) 71%
It was also concerning that the Beta variant that was first identified in Africa was starting to increase in numbers especially in the Southern provinces in Thailand.
There is currently no data with regards to the behavior of each new emerging Delta variant sub-species or second generation variants with regards to the efficacy of the various current COVID-19 vaccines against them but a latest Japanese study warns that the Delta variant is evolving towards developing total vaccine resistance. https://www.thailandmedical.news/news/warning-the-delta-variant-is-mutating-rapidly,-sri-lanka-reports-4-new-delta-variants,-japanese-study-warns-about-vaccine-efficacy-and-ade-issues
Health authorities in Sri Lanka’s reported on Monday the Delta variant of the SARS-CoV-2 coronavirus which is spreading across the country has acquired four mutations due to the high transmission rate.
Professor Dr Neelika Malavige, head of the department of immunology and molecular sciences at the Sri Jayawardenapura University told Thailand Medical News, “We discovered the 4 new Delta variants following gene sequencings of samples taken from a number of infected patients. One of the Delta mutations (A-222V) is seen in many countries, another (A-1078S) is found in Sri Lanka and Malaysia, while the other two (A-701S and R-24C) are only found in Sri Lanka. These possess unique mutations in the Delta variant genome but do not make these viruses new variants. Some of these mutations appear concerning and we have reported the discovery to the WHO and also U.S. CDC.”
Dr Malavige added, “We had identified many other mutations in the previous Alpha variant and in our variant of the Sri Lankan lineage (responsible for the second wave), which were of no significance. Some of the Delta variant viruses seen in Sri Lanka might have certain unique concerning mutations and more detailed research is warranted.”
At the moment that there is no evidence yet whether these new mutations found in the Delta variant in Sri Lanka will affected COVID-19 efficacy he added.
The South-East Asian nation is presently facing a rising wave of COVID-19 infections, suspected to be caused by the Delta variant with authorities declaring a nationwide quarantine curfew since last Friday. Hospitals are exhausted with rising admissions while oxygen dependency has also risen among the patients.
The country has recorded over 7,000 deaths and 390,000 COVID-19 infections.
Australia had also reported the emergence of a new Delta variant that is not only fast spreading but some researchers claim are able to cause disease severity as a certain unique ORF7a deletion found on it and is thought to promote cell fusion.
The SARS-CoV-2 coronavirus encodes proteins that modulate antiviral responses (ORF3b, ORF6, ORF7a), including reducing type 1 interferon. The ORF7a of SARS-CoV-2 is an ortholog of the corresponding SARS-CoV antagonist of host restriction factor BST-2/CD317/Tetherin that induces apoptosis.
Most importantly cells with reduced BST2 enhance SARS-CoV-2 replication thereby releasing more virus and indirectly increases viral shedding loads, thus helping to spread the disease even more.
The study findings were published on a preprint server and are currently being peer reviewed. https://www.medrxiv.org/content/10.1101/2021.08.18.21262089v1
Detailed analysis of the index case reveals a sub-consensus level of sequencing reads (~25%) that support a 17-nucleotide deletion in ORF7a (ORF7aΔ17del). ORF7aΔ17del induces a frameshift mutation in ORF7a, which truncates the peptide and potentially leads to reduced suppression of host restriction factor BST-2/CD317/Tetherin.
Despite this, the mutation has rapidly become represented at the consensus level in subsequent cases: approximately 72% of SARSCoV-2 genomes in the Australian outbreak possess ORF7aΔ17del, and 99.7% (1534/1538) of Delta genomes on GISAID with ORF7aΔ17del originate from the current Australian outbreak (5 August 2021).
Alarmingly the global abundance of this mutation might be underestimated given the difficulty of variant calling software correctly calling insertion/deletions (indels), the common inability of phylogenetics software to take indels into account, and the tendency of GISAID to not release submissions that contain a frameshift mutation (unless specifically requested).
On the whole, the rapid increase of persistent ORF7aΔ17del variants is concerning, and suggests either a chance founder effect with a neutral mutation yet to be purged, or that the ORF7aΔ17del mutation provides a direct selective advantage.
Malaysia
Malaysia Surges Not Caused By Delta But Rather By Its Unique Variants Called B.1.524 and AU.2 Variants In Malaysia Which are More Transmissible Than Delta Variant And Alarmingly A New Mutation G1223C Is Appearing In Many Lineages! ng>
Malaysia is witnessing high COVID-19 infections rates in the last few months and despite an intensive vaccination programme, it has to date only managed to get about 36.8% of its population of about 32.83 million fully vaccinated.
However Malaysia’s COVID-19 crisis is slightly different from neighboring Thailand that is seriously inflicted with various VOCs (Variants of Concern) especially the Delta variant. The Alpha and Beta variants have also been found in the Thailand and God knows what else that the local authorities are not revealing. Thailand is already being bestowed the title of the “Hub of SARS-CoV-2 Variants!”
In the case of Malaysia, the surge is being driven by two unique variants called the B.1.524 and AU.2 variants.
Malaysian researchers from the Universiti Malaysia Pahang and the International Islamic University of Malaysia were the ones who identified the variants in Malaysia. It should also be noted that these variants have also been detected already in Thailand as well from spread via Malaysia.
The study team aimed to report new Malaysian lineages responsible for the sustained spikes in COVID-19 cases during the third wave of the pandemic.
Patients whose nasopharyngeal and oropharyngeal swabs were confirmed positive by real-time RT-PCR with Ct-value < 25 were chosen for WGS(Whole Genome Sequencing). The 10 SARS-CoV-2 isolates obtained were then sequenced, characterized and analyzed, including 1356 sequences of the dominant lineages of D614G variant currently circulating throughout Malaysia.
Interestingly the prevalence of clade GH and G formed strong ground of the discovery of two Malaysian lineages that caused sustained spikes of cases locally ie the B.1.524 and AU.2 variants.
Statistical analysis on the association of gender and age group with Malaysian lineages revealed a significant association (p < 0.05). Phylogenetic analysis revealed dispersion of 41 lineages, for which 22 lineages are still active.
Importantly detailed mutational analysis revealed a unique G1223C missense mutation in Transmembrane Domain of Spike protein. Thus, calls for the large-scale WGS analysis of strains found around the world for greater understanding of viral evolution and genetic diversity especially in addressing the question of the effect of deleterious substitution mutation in transmembrane region of Spike protein.
The study findings were also published on a preprint server and are currently also being peer-reviewed. https://www.medrxiv.org/content/10.1101/2021.08.11.21261902v1
Interesting observations from the study were that these two variants possibly are even more transmissible than the Delta variant.
The most important finding from the study was the discovery of the present of an emerging mutation found on all the new variants in Malaysia ie the G1223C mutation.
While the significance of G1223C mutation is still unknown, it is well known that Spike protein mediates entry of SARS-CoV-2 into target cells through two steps. First, it involves binding of RBD to its receptor human ACE2 and is proteolytically activated by human proteases at the S1/S2 boundary. Second, it follows by S2 of which include TM domain will undergo structural change to mediate viral membrane fusion with targeted cells.
To date, very little attention was put on the TM domain in the requirements for SARS-CoV cell entry. Although sequence analysis on TM domain among all coronaviruses Spike protein conducted previously revealed a high conservation rate, extensive mutation in TM domain of SARS-CoV however caused incapability of the virus to establish complete membrane fusion process.
Highly conserved small amino acids in TM domain of SARS-CoV-2 Spike protein (G1219, A1222, G1223, A1226) which initially thought to be important for TM domain oligomerization, but latest finding showed neither glycine nor alanine in the trimer structure appear to be important for hydrophobic core formation. Thus, suggesting a possible role of the glycine motif is in a later step of fusion.
The study team believes the effect of G1223C mutation in TM domain deserves to further analyze in future functional experiments for addressing above question.
If the G1223C mutation does promote fusion, it could alarmingly imply that variants with these mutations could cause more severe conditions in those infected!
South Africa Reports Emergence Of New Worrisome SARS-CoV-2 Variant C.1.2 That Has Enhanced Transmissibility And Immune Evasion!
South African researchers from the National Institute for Communicable Diseases (NICD), University of the Witwatersrand, University of Cape Town, University of Pretoria, University of KwaZulu-Natal and the Charlotte Maxeke Johannesburg Academic Hospital have discovered a new emerged SARS-CoV-2 variant called C.1.2 that is currently gaining dominance in circulation and could change the course of the COVID-19 pandemic as it is far more transmissible and also immune evasive due to enhancement by newer mutations found on its genome.
Both the WHO and also the U.S. CDC has been informed of the development and from the GSAID platforms it can be seen that C.1.2 variants has since been detected across the majority of the provinces in South Africa and alarmingly also in seven other countries spanning Africa, Europe, Asia and Oceania!
The report from study findings were published on a preprint server and are currently being peer reviewed. https://www.medrxiv.org/content/10.1101/2021.08.20.21262342v1
Various emerging SARS-CoV-2 variants of interest have been associated with increased transmissibility, neutralization resistance and disease severity.
Fortunately ongoing SARS-CoV-2 genomic surveillance world-wide has improved researcher’s ability to rapidly identify such variants.
The South African study team reports the identification of a potential variant of interest assigned to the PANGO lineage C.1.2. This lineage was first identified in May 2021 and evolved from C.1, one of the lineages that dominated the first wave of SARS-CoV-2 infections in South Africa and was last detected in January 2021.
Alarmingly, the emergence of C.1.2 variant was associated with an increased substitution rate, as was previously observed with the emergence of the Alpha, Beta and Gamma variants of concern (VOCs).
While the VOI Lambda (C.37) is phylogenetically closest to C.1.2, the latter has distinct lineage-defining mutations.
The new C.1.2 variant contains multiple substitutions (R190S, D215G, N484K, N501Y, H655Y and T859N) and deletions (Y144del, L242-A243del) within the spike protein, which have been observed in other VOCs and are associated with increased transmissibility and reduced neutralization sensitivity.
However, of greater concern is the accumulation of additional mutations (C136F, Y449H and N679K) which are also likely to impact neutralization sensitivity or furin cleavage and therefore replicative fitness.
Though these mutations occur in the majority of C.1.2 viruses, there is additional variation within the spike region of this lineage, suggesting ongoing intralineage evolution.
Approximately 44% of the viruses also contain a P25L mutation in the NTD, ~19% have L585F in S1, ~16% have T478K in the RBM, ~11% contain P681H adjacent to the furin cleavage site, 8% have D936H, and a further ~8% have H1101Q in S2. The majority of these mutations (P9L, C136F, R190S, D215G, L242del, A243del, Y449H, E484K, N501Y, H655Y, and T716I) appeared together early in the lineage evolution (Fig. 3a). Thereafter, the majority of sequences have also accumulated the mutations Y144del, N679K and T859N. The mutations P25L, W152R, R 346K, T478K, L585F, N440K, P681H, A879T, D936H and H1101Q can be seen in some of the smaller clusters from more recent sequences, further highlighting continued evolution within the lineage.
Researchers are worried that continued evolution of this lineage could result in the much anticipated deadly ‘Omega’ variant.
The C.1.2 lineage was first detected in the Mpumalanga and Gauteng provinces of South Africa, in May 2021. In June 2021, it was also detected in the KwaZulu-Natal and Limpopo provinces of South Africa as well as in England and China.
As of August 13, 2021 the C.1.2 lineage has been detected in 6/9 South African provinces (including the Eastern Cape and Western Cape), the Democratic Republic of the Congo (DRC), Mauritius, New Zealand, Portugal and Switzerland
The study team warns that while the phenotypic characteristics and epidemiology of C.1.2 are being defined, it is important to highlight this lineage given its concerning constellations of mutations.
reference link:
- https://www.cdc.gov/coronavirus/2019-ncov/variants/delta-variant.html



{kind=link}