











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



Il 27 luglio 2021, il CDC ha pubblicato una guida aggiornata sulla necessità di aumentare urgentemente la copertura vaccinale COVID-19 e una raccomandazione per tutti coloro che si trovano in aree a trasmissione sostanziale o alta di indossare una maschera in luoghi pubblici al chiuso, anche se sono completamente vaccinati.
CDC ha emesso questa nuova guida a causa di diversi sviluppi riguardanti e segnali di dati emergenti di recente.
- Alla fine di giugno, la nostra media mobile a 7 giorni dei casi segnalati era di circa 12.000. Il 27 luglio, la media mobile a 7 giorni dei casi ha raggiunto oltre 60.000. Questo tasso di casi sembrava più simile al tasso di casi che avevamo visto prima che il vaccino fosse ampiamente disponibile.
In secondo luogo, sono emersi nuovi dati secondo cui la variante Delta era più infettiva e stava portando a una maggiore trasmissibilità rispetto ad altre varianti, anche negli individui vaccinati. Ciò include dati pubblicati di recente da CDC e dai nostri partner per la salute pubblica, dati di sorveglianza non pubblicati che saranno disponibili pubblicamente nelle prossime settimane, informazioni incluse nel Science Brief aggiornato sui vaccini e le vaccinazioni COVID-19 del CDC e le indagini sull’epidemia in corso legate alla variante Delta .
Delta è attualmente il ceppo predominante del virus negli Stati Uniti. Di seguito è riportato un riepilogo di alto livello di ciò che gli scienziati del CDC hanno recentemente appreso sulla variante Delta. Maggiori informazioni saranno rese disponibili quando più dati saranno pubblicati o rilasciati in altri formati.
Infezioni e diffusione
- La variante Delta è più contagiosa: la variante Delta è altamente contagiosa, più di 2 volte più contagiosa delle varianti precedenti.
- Alcuni dati suggeriscono che la variante Delta potrebbe causare malattie più gravi rispetto ai ceppi precedenti in persone non vaccinate. In due diversi studi condotti in Canada e in Scozia, i pazienti infettati con la variante Delta avevano maggiori probabilità di essere ricoverati in ospedale rispetto ai pazienti infetti da Alpha o dai ceppi virali originali.
- Le persone non vaccinate rimangono la preoccupazione maggiore: sebbene le infezioni da esordio si verifichino molto meno spesso rispetto alle infezioni tra le persone non vaccinate, gli individui infettati dalla variante Delta, comprese le persone completamente vaccinate con infezioni sintomatiche, possono trasmetterla ad altri. Il CDC sta continuando a valutare i dati sulla trasmissione di persone completamente vaccinate con infezioni asintomatiche. Tuttavia, il rischio maggiore di trasmissione è tra le persone non vaccinate che hanno molte più probabilità di contrarre e quindi trasmettere il virus.
- Le persone completamente vaccinate con infezioni rivoluzionarie della variante Delta possono diffondere il virus ad altri. Tuttavia, le persone vaccinate sembrano essere infettive per un periodo più breve: le varianti precedenti in genere producevano meno virus nel corpo delle persone infette completamente vaccinate (infezioni rivoluzionarie) rispetto alle persone non vaccinate. Al contrario, la variante Delta sembra produrre la stessa elevata quantità di virus sia nelle persone non vaccinate che in quelle completamente vaccinate. Tuttavia, come altre varianti, anche la quantità di virus prodotta dalle infezioni rivoluzionarie Delta nelle persone completamente vaccinate diminuisce più rapidamente rispetto alle infezioni nelle persone non vaccinate. Ciò significa che le persone completamente vaccinate sono probabilmente infettive per meno tempo rispetto alle persone non vaccinate.

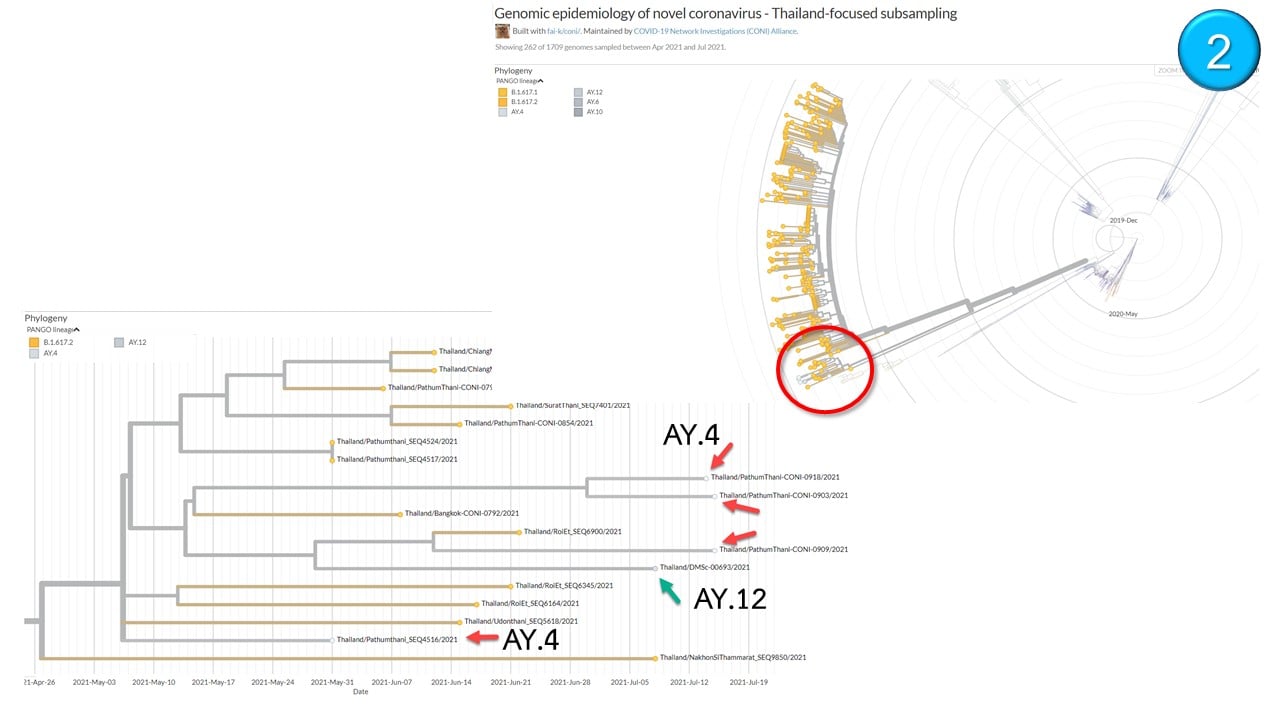
Il Centro thailandese per la genomica medica ha annunciato che finora in Thailandia sono state identificate 4 diverse varianti delta di seconda generazione con mutazioni uniche in 14 individui diversi.
Secondo gli esperti thailandesi locali, la variante Delta AY.4 è stata trovata in un totale di 9 casi di sequenziamento genico in Thailandia con la provincia di Pathumthani che ha riportato 4 casi e un caso nelle seguenti province: Buriram, Kamphaeng Phet, Chiang Mai, Samut Prakan e Chonburi.
A Bangkok, è stato rilevato un caso ciascuna delle varianti AY.6, AY.10 e AY.12 Delta mentre la provincia di Surat Thani ha riportato 2 casi della variante AY.12 .
Credito: Thailand Centre For Medical Genomics
Ad oggi, a livello globale, nelle ultime 96 ore sono state identificate un totale di 27 diverse varianti Delta di seconda e terza generazione. 22 di questi sono nominati come sottospecie Delta da AY.1 a 22 mentre il resto è ancora in fase di studio. Nella nomenclatura di denominazione la sottospecie della variante Delta (B.1.617.2) AY.1 sarebbe etichettata come B.1.617.2.1
I ricercatori globali sono preoccupati perché alcune di queste varianti sono in possesso di mutazioni su di esse e sono in corso studi più dettagliati per capire come queste mutazioni potrebbero causare differenze nei modelli di patogenesi e oltre a migliorare l’idoneità virale, la virulenza e anche la trasmissibilità.
Le autorità locali thailandesi hanno anche riferito che l’attuale predominanza dei vari COV o varianti preoccupanti nel paese è la seguente:
B.1.1.7 (alpha) 11%
B.1.351 (Beta) 14%
B.1.617.2 (Delta) 71%
Era anche preoccupante che la variante Beta identificata per la prima volta in Africa stesse iniziando ad aumentare di numero, specialmente nelle province meridionali della Thailandia.
Al momento non ci sono dati sul comportamento di ogni nuova sottospecie emergente di variante Delta o varianti di seconda generazione per quanto riguarda l’efficacia dei vari attuali vaccini COVID-19 contro di loro, ma un ultimo studio giapponese avverte che la variante Delta si sta evolvendo verso lo sviluppo di una resistenza totale ai vaccini. https://www.thailandmedical.news/news/warning-the-delta-variant-is-mutating-rapidly,-sri-lanka-reports-4-new-delta-variants,-japanese-study-warns-about-vaccine-efficacy-and-ade-issues
Le autorità sanitarie dello Sri Lanka hanno riferito lunedì che la variante Delta del coronavirus SARS-CoV-2 che si sta diffondendo in tutto il paese ha acquisito quattro mutazioni a causa dell’elevata velocità di trasmissione.
La professoressa Dr Neelika Malavige, capo del dipartimento di immunologia e scienze molecolari presso l’Università Sri Jayawardenapura, ha dichiarato a Thailand Medical News: “Abbiamo scoperto le 4 nuove varianti Delta in seguito al sequenziamento genico di campioni prelevati da un certo numero di pazienti infetti. Una delle mutazioni Delta (A-222V) è presente in molti paesi, un’altra (A-1078S) si trova in Sri Lanka e Malesia, mentre le altre due (A-701S e R-24C) si trovano solo in Sri Lanka. Questi possiedono mutazioni uniche nel genoma della variante Delta, ma non rendono questi virus nuove varianti. Alcune di queste mutazioni sembrano preoccupanti e abbiamo segnalato la scoperta all’OMS e anche al CDC degli Stati Uniti .
Il dottor Malavige ha aggiunto: “Avevamo identificato molte altre mutazioni nella precedente variante Alpha e nella nostra variante del lignaggio dello Sri Lanka (responsabile della seconda ondata), che non avevano alcun significato. Alcuni dei virus della variante Delta visti in Sri Lanka potrebbero avere alcune mutazioni uniche e sono giustificate ricerche più dettagliate.
Al momento non ci sono ancora prove se queste nuove mutazioni trovate nella variante Delta in Sri Lanka influenzeranno l’efficacia del COVID-19, ha aggiunto.
La nazione del sud-est asiatico sta attualmente affrontando un’ondata crescente di infezioni da COVID-19, sospettate di essere causate dalla variante Delta con le autorità che hanno dichiarato un coprifuoco di quarantena a livello nazionale dallo scorso venerdì. Gli ospedali sono esausti con l’aumento dei ricoveri, mentre è aumentata anche la dipendenza dall’ossigeno tra i pazienti.
Il paese ha registrato oltre 7.000 morti e 390.000 infezioni da COVID-19.
L’Australia ha anche segnalato l’emergere di una nuova variante Delta che non solo si sta diffondendo rapidamente, ma alcuni ricercatori affermano che sono in grado di causare la gravità della malattia come una certa delezione unica di ORF7a trovata su di essa e si pensa che promuova la fusione cellulare.
Il coronavirus SARS-CoV-2 codifica per proteine che modulano le risposte antivirali (ORF3b, ORF6, ORF7a), inclusa la riduzione dell’interferone di tipo 1. L’ ORF7a di SARS-CoV-2 è un ortologo del corrispondente antagonista SARS-CoV del fattore di restrizione dell’ospite BST-2/CD317/Tetherin che induce l’apoptosi.
Ancora più importante, le cellule con BST2 ridotto migliorano la replicazione di SARS-CoV-2, rilasciando così più virus e aumentando indirettamente i carichi di diffusione virale, contribuendo così a diffondere ancora di più la malattia.
I risultati dello studio sono stati pubblicati su un server di prestampa e sono attualmente in fase di revisione paritaria. https://www.medrxiv.org/content/10.1101/2021.08.18.21262089v1
L’analisi dettagliata del caso indice rivela un livello di subconsenso di letture di sequenziamento (~ 25%) che supportano una delezione di 17 nucleotidi in ORF7a (ORF7aΔ17del). ORF7aΔ17del induce una mutazione frameshift in ORF7a, che tronca il peptide e potenzialmente porta a una ridotta soppressione del fattore di restrizione dell’ospite BST-2/CD317/Tetherin.
Nonostante ciò, la mutazione è stata rapidamente rappresentata a livello di consenso nei casi successivi: circa il 72% dei genomi SARSCoV-2 nell’epidemia australiana possiede ORF7aΔ17del e il 99,7% (1534/1538) dei genomi Delta su GISAID con ORF7aΔ17del ha origine dal attuale focolaio australiano (5 agosto 2021).
In modo allarmante, l’abbondanza globale di questa mutazione potrebbe essere sottovalutata, data la difficoltà di chiamare le varianti software che chiamano correttamente inserimenti/cancellazioni (indel), la comune incapacità dei software di filogenetica di prendere in considerazione gli indel e la tendenza di GISAID a non rilasciare contributi che contengono un mutazione frameshift (se non espressamente richiesto).
Nel complesso, il rapido aumento delle varianti ORF7aΔ17del persistenti è preoccupante e suggerisce un effetto fondatore casuale con una mutazione neutra ancora da eliminare, o che la mutazione ORF7aΔ17del fornisce un vantaggio selettivo diretto.
Malaysia
Le impennate della Malesia non sono causate da Delta, ma piuttosto dalle sue varianti uniche chiamate varianti B.1.524 e AU.2 in Malesia che sono più trasmissibili della variante Delta e una nuova mutazione G1223C sta apparendo in modo allarmante in molti lignaggi! ng> La
Malesia sta assistendo ad alti tassi di infezione da COVID-19 negli ultimi mesi e nonostante un intenso programma di vaccinazione, ad oggi è riuscita a vaccinare solo circa il 36,8% della sua popolazione di circa 32,83 milioni.
Tuttavia, la crisi COVID-19 della Malesia è leggermente diversa dalla vicina Thailandia, gravemente inflitta da vari VOC (Variant of Concern), in particolare la variante Delta. Le varianti Alpha e Beta sono state trovate anche in Thailandia e chissà cos’altro che le autorità locali non stiano rivelando. Alla Thailandia è già stato conferito il titolo di “Hub of SARS-CoV-2 Variants!”
Nel caso della Malesia, l’aumento è guidato da due varianti uniche chiamate varianti B.1.524 e AU.2 .
I ricercatori malesi dell’Universti Malaysia Pahang e dell’International Islamic University of Malaysia sono stati quelli che hanno identificato le varianti in Malaysia. Va inoltre notato che queste varianti sono state rilevate anche già in Thailandia e dalla diffusione attraverso la Malesia.
Il team di studio mirava a segnalare nuovi lignaggi malesi responsabili dei picchi sostenuti nei casi di COVID-19 durante la terza ondata della pandemia.
I pazienti i cui tamponi nasofaringei e orofaringei sono stati confermati positivi dalla RT-PCR in tempo reale con valore Ct < 25 sono stati scelti per il WGS (Whole Genome Sequencing). I 10 isolati SARS-CoV-2 ottenuti sono stati quindi sequenziati, caratterizzati e analizzati, incluse 1356 sequenze dei lignaggi dominanti della variante D614G attualmente in circolazione in tutta la Malesia.
È interessante notare che la prevalenza di clade GH e G ha costituito una solida base per la scoperta di due lignaggi malesi che hanno causato picchi sostenuti di casi localmente, ad esempio le varianti B.1.524 e AU.2.
L’analisi statistica sull’associazione del genere e della fascia di età con i lignaggi malesi ha rivelato un’associazione significativa ( p <0,05). L’analisi filogenetica ha rivelato la dispersione di 41 lignaggi, per i quali 22 lignaggi sono ancora attivi.
Un’analisi mutazionale particolarmente dettagliata ha rivelato una mutazione missenso G1223C unica nel dominio transmembrana della proteina Spike. Pertanto, richiede l’analisi WGS su larga scala dei ceppi trovati in tutto il mondo per una maggiore comprensione dell’evoluzione virale e della diversità genetica, in particolare nell’affrontare la questione dell’effetto della mutazione sostitutiva deleteria nella regione transmembrana della proteina Spike.
I risultati dello studio sono stati anche pubblicati su un server di prestampa e sono attualmente in fase di revisione paritaria. https://www.medrxiv.org/content/10.1101/2021.08.11.21261902v1
Le osservazioni interessanti dello studio sono state che queste due varianti sono forse ancora più trasmissibili della variante Delta.
La scoperta più importante dello studio è stata la scoperta del presente di una mutazione emergente trovata su tutte le nuove varianti in Malesia, ovvero la mutazione G1223C.
Sebbene il significato della mutazione G1223C sia ancora sconosciuto, è ben noto che la proteina Spike media l’ingresso di SARS-CoV-2 nelle cellule bersaglio attraverso due passaggi. Innanzitutto, implica il legame di RBD al suo recettore ACE2 umano ed è attivato proteoliticamente dalle proteasi umane al confine S1/S2. In secondo luogo, ne consegue che S2 include il dominio TM che subirà un cambiamento strutturale per mediare la fusione della membrana virale con le cellule mirate.
Ad oggi, è stata posta pochissima attenzione sul dominio TM nei requisiti per l’ingresso delle cellule SARS-CoV. Sebbene l’analisi della sequenza sul dominio TM tra tutti i coronavirus La proteina Spike condotta in precedenza abbia rivelato un alto tasso di conservazione, un’ampia mutazione nel dominio TM di SARS-CoV ha tuttavia causato l’incapacità del virus di stabilire un completo processo di fusione della membrana.
Piccoli amminoacidi altamente conservati nel dominio TM della proteina Spike SARS-CoV-2 (G1219, A1222, G1223, A1226) che inizialmente si pensava fosse importante per l’oligomerizzazione del dominio TM, ma l’ultima scoperta ha mostrato che né la glicina né l’alanina nella struttura del trimero sembrano essere importante per la formazione del nucleo idrofobo. Quindi, suggerendo un possibile ruolo del motivo glicina è in una fase successiva della fusione.
Il team di studio ritiene che l’effetto della mutazione G1223C nel dominio TM meriti di essere ulteriormente analizzato in futuri esperimenti funzionali per affrontare la domanda di cui sopra.
Se la mutazione G1223C promuove la fusione, potrebbe implicare in modo allarmante che le varianti con queste mutazioni potrebbero causare condizioni più gravi nelle persone infette!
Il Sudafrica segnala l’emergere di una nuova preoccupante variante SARS-CoV-2 C.1.2 che ha migliorato la trasmissibilità e l’evasione immunitaria!
Ricercatori sudafricani del National Institute for Communicable Diseases (NICD), University of the Witwatersrand, University of Cape Town, University of Pretoria, University of KwaZulu-Natal e Charlotte Maxeke Johannesburg Academic Hospital hanno scoperto un nuovo SARS-CoV-2 emerso variante chiamata C.1.2 che sta attualmente guadagnando il dominio in circolazione e potrebbe cambiare il corso della pandemia di COVID-19 in quanto è molto più trasmissibile e anche immunitaria evasiva a causa del potenziamento di nuove mutazioni trovate sul suo genoma.
Sia l’OMS che il CDC degli Stati Uniti sono stati informati dello sviluppo e dalle piattaforme GSAID si può vedere che varianti C.1.2 sono state rilevate da allora nella maggior parte delle province del Sud Africa e in modo allarmante anche in altri sette paesi dell’Africa , Europa, Asia e Oceania!
Il rapporto dei risultati dello studio è stato pubblicato su un server di prestampa e attualmente è in fase di revisione paritaria. https://www.medrxiv.org/content/10.1101/2021.08.20.21262342v1
Varie varianti emergenti di SARS-CoV-2 di interesse sono state associate ad una maggiore trasmissibilità, resistenza alla neutralizzazione e gravità della malattia.
Fortunatamente la sorveglianza genomica SARS-CoV-2 in corso in tutto il mondo ha migliorato la capacità dei ricercatori di identificare rapidamente tali varianti.
Il gruppo di studio sudafricano riporta l’identificazione di una potenziale variante di interesse assegnata al ceppo PANGO C.1.2. Questo lignaggio è stato identificato per la prima volta nel maggio 2021 e si è evoluto da C.1, uno dei lignaggi che ha dominato la prima ondata di infezioni da SARS-CoV-2 in Sud Africa ed è stato rilevato l’ultima volta nel gennaio 2021. In modo
allarmante, l’emergere di C.1.2 La variante è stata associata a un aumento del tasso di sostituzione, come precedentemente osservato con l’emergere delle varianti di preoccupazione (VOC) Alfa, Beta e Gamma.
Mentre il VOI Lambda (C.37) è filogeneticamente più vicino a C.1.2, quest’ultimo ha mutazioni che definiscono il lignaggio distinte.
La nuova variante C.1.2 contiene più sostituzioni (R190S, D215G, N484K, N501Y, H655Y e T859N) e delezioni (Y144del, L242-A243del) all’interno della proteina spike, che sono state osservate in altri COV e sono associate ad una maggiore trasmissibilità e ridotta sensibilità alla neutralizzazione.
Tuttavia, di maggiore preoccupazione è l’accumulo di ulteriori mutazioni (C136F, Y449H e N679K) che possono anche avere un impatto sulla sensibilità alla neutralizzazione o sulla scissione della furina e quindi sull’idoneità replicativa.
Sebbene queste mutazioni si verifichino nella maggior parte dei virus C.1.2, vi è un’ulteriore variazione all’interno della regione del picco di questo lignaggio, suggerendo un’evoluzione intralineare in corso.
Circa il 44% dei virus contiene anche una mutazione P25L nel NTD, circa il 19% ha L585F in S1, circa il 16% ha T478K nell’RBM, circa l’11% contiene P681H adiacente al sito di scissione della furina, l’8% ha D936H e un ulteriore ~8% ha H1101Q in S2. La maggior parte di queste mutazioni (P9L, C136F, R190S, D215G, L242del, A243del, Y449H, E484K, N501Y, H655Y e T716I) sono apparse insieme all’inizio dell’evoluzione del lignaggio (Fig. 3a). Successivamente, la maggior parte delle sequenze ha anche accumulato le mutazioni Y144del, N679K e T859N. Le mutazioni P25L, W152R, R 346K, T478K, L585F, N440K, P681H, A879T, D936H e H1101Q possono essere osservate in alcuni dei cluster più piccoli di sequenze più recenti, evidenziando ulteriormente l’evoluzione continua all’interno del lignaggio.
I ricercatori sono preoccupati che la continua evoluzione di questo lignaggio possa portare alla tanto attesa variante mortale “Omega”.
Il lignaggio C.1.2 è stato rilevato per la prima volta nelle province di Mpumalanga e Gauteng in Sudafrica, nel maggio 2021. Nel giugno 2021, è stato rilevato anche nelle province di KwaZulu-Natal e Limpopo in Sudafrica, nonché in Inghilterra e Cina.
A partire dal 13 ago 2021 il lignaggio C.1.2 è stato rilevato in 6/9 province del sud africani (tra cui l’Eastern Cape e Western Cape), la Repubblica democratica del Congo (RDC), Mauritius, Nuova Zelanda, Portogallo e Svizzera
Il il team di studio avverte che mentre si stanno definendo le caratteristiche fenotipiche e l’epidemiologia di C.1.2, è importante evidenziare questo lignaggio date le sue preoccupanti costellazioni di mutazioni.
collegamento di riferimento:
- https://www.cdc.gov/coronavirus/2019-ncov/variants/delta-variant.html



{kind=link}