











 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



New alarming study finding on the multitude of negative effects that the novel coronavirus has on the human host, this time from a research by scientist from Yale University School of Medicine, Icahn School of Medicine at Mount Sinai, and Iowa State University.
The research findings reveals that the SARS-CoV-2 coronavirus expresses a microRNA or miRNA called vmiR-5p that is able to reduce and disrupt human host transcription, thus suppressing selected host genes which aids I the virus pathogenicity.
The study team investigated the impact of SARS-CoV-2 infection on host microRNA (miRNA) populations in three human lung-derived cell lines, as well as in nasopharyngeal swabs from SARS-CoV-2 infected individuals.
The team did not detect any major and consistent differences in host miRNA levels after SARS-CoV-2 infection.
Shockingly instead, the study team unexpectedly discovered a viral miRNA-like small RNA, named vmiR-5p (for viral miRNA), derived from the SARS-CoV-2 ORF7a transcript. Its abundance ranges from low to moderate as compared to host miRNAs.
This vmiR-5p miRNA functionally associates with Argonaute proteins which are core components of the RNA interference pathway that lead to downregulation of host transcripts.
It was found that one such host messenger RNA that was suppressed, encodes Basic Leucine Zipper ATF-Like Transcription Factor 2 (BATF2), which is linked to interferon signaling.
The study findings demonstrate that vmiR-5p production relies on cellular machinery, yet is independent of Drosha protein, and is enhanced by the presence of a strong and evolutionarily conserved hairpin formed within the ORF7a sequence.
This newly discovered viral miRNA called vmiR-5p may contribute to SARS-CoV-2 pathogenesis.
The study findings were published on a preprint server and are currently being peer reviewed. https://www.biorxiv.org/content/10.1101/2021.09.08.459464v1
The SARS-CoV-2 coronavirus that triggered the ongoing COVID-19) pandemic that brought life to a grinding halt in much of the inhabited world for most of 2020 and 2021 is still fast accelerating with major surges expected in coming weeks.
As these surges continue to cause thousands of new infections and deaths worldwide each day, researchers are earnestly seeking to understand how SARS-CoV-2 works to infect host cells and inactivate host defenses.
This Medical Alert warns about the new study that finds the viral non-coding ribonucleic acid (RNA) dubbed vmiR-5p, derived from the viral gene open reading frame 7a (ORF7a) appears to reduce host transcription, which could contribute to the pathogenicity of SARS-CoV-2. This new finding could provide a new therapeutic target against SARS-CoV-2.
The SARS-CoV-2 coronavirus is a large RNA virus that causes both human and animal infection. Although SARS-CoV-2 generally is associated with its respiratory manifestations, in severe and critical cases, it induces overwhelming inflammation that results in multi-organ dysfunction and even deat h.
The present study involves microRNAs (miRNAs), which are about 22 nucleotides in length. These miRNAs comprise non-coding RNA molecules (ncRNAs) that modulate gene expression in the post-transcriptional stage. This action is mediated by partial inhibition or mRNA decay.
Although messenger RNA (mRNA) is typically being synthesized in the cytoplasm, the enzyme RNA polymerase II causes primary miRNA molecules to be transcribed, all of which have a characteristic tendency to form strong hairpins. The endonuclease Drosha recognizes such hairpin sequences and cleaves them, leading to the production of miRNA precursors that are about 70 nucleotides in length.
Subsequently these precursor molecules are trafficked into the cytoplasm where they are cleaved by another enzyme known as Dicer, which assists in the formation of duplex miRNAs that are 22 nucleotides in length. These become the cargo of host Argonaute (Ago) proteins.
It should be noted that at this point, the passenger strand is removed. The Ago proteins bind to target sequences that are partly complementary to the miRNA. These sequences are mostly in the 3′ untranslated region (UTR) of mRNAs. This binding causes inhibition of translation of these mRNAs or their decay.
Importantly when the target mRNA and binding miRNA sequences are almost completely complementary, the Ago2 protein cleaves the mRNA. This cleavage is an action that is more characteristic of small interfering RNA molecules (siRNAs).
Again, it should be noted that both these functions are part of the RNA interference (RNAi) pathway.
The Ago2-mediated RNA cleavage however occurs at a lower copy number and silences the target gene more powerfully than the standard miRNA mode of action.
Interestingly the number and types of miRNAs differ by the host cell and stage of development. Also, each miRNA is capable of reducing the level of expression of a range of transcripts. The outcome is a precise tuning of gene expression of almost all mRNAs as required by each type of cell at its particular stage.
Importantly the presence of abnormal miRNAs is typically a marker of disease. Very often, this is the case with a viral infection, as viruses take over this mechanism to reduce the level of host miRNAs or produce their own miRNAs.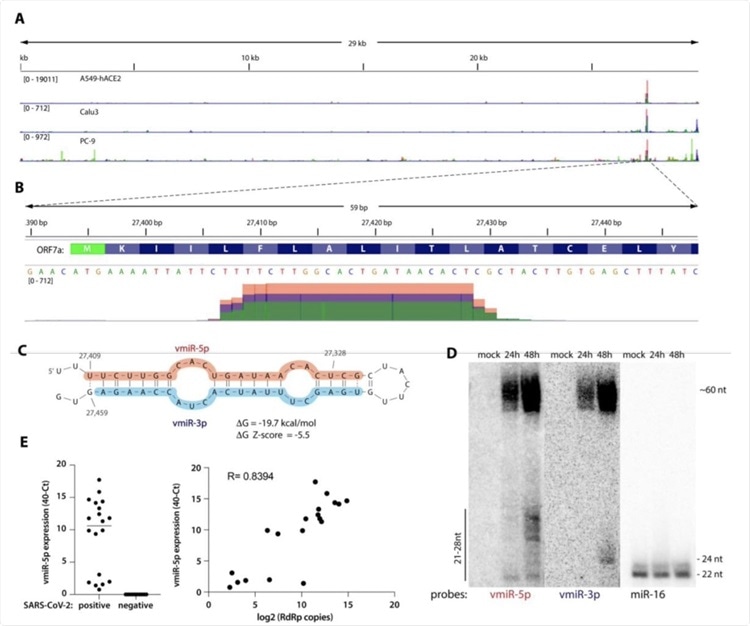
A) Viral small RNA reads obtained from the three cell lines infected with SARS-CoV-2 map to a single distinct peak within the viral genome (data for MOI 5, 24 hpi are shown). The replicates for each cell line were overlayed on a single track (represented by different colors) and are normalized to 107 total reads. MOI-multiplicity of infection. B) The reads coming from SARS-CoV-2 (∼20 nt-long) map near the beginning of the ORF7a gene (encoded amino acids are shown above the nucleotide sequence). Data from Calu-3 cells are shown. C) vmiR-5p forms a hairpin with the sequence immediately downstream in the viral genome. Shaded nucleotides indicate the sequences detected by Northern blot probes: pink for 5p and blue for 3p. D) vmiR-5p can be detected by Northern blotting of extracts from Calu-3 cells infected with SARS-CoV-2 at MOI 0.05. E) As measured by custom TaqMan RT-qPCR, vmiR-5p is present in nasopharyngeal samples from SARS-CoV-2-infected individuals (right panel), with its levels correlating with viral load (left panel). RdRp ie RNA-dependent RNA polymerase
For instance, herpes viruses transcribes certain sequences that lead to the selective decay of some host miRNAs through a process known as target-directed miRNA degradation (TDMD).
Another example is the poxvirus poly(A) polymerase that causes extensive miRNA polyadenylation that ultimately leads to miRNA decay.
Also viruses can use unconventional non-RNAi pathways to avoid the need for Drosha, rather than synthesizing their own miRNAs in the cytoplasm.
It should be noted that coronaviruses such as SARS-CoV-2 may be of this kind, producing small viral RNAs (svRNAs) as part of their disease-producing activity, including miRNA-like strands that trigger inflammation and type I interferon (IFN) signaling.
This study findings reports the discovery of a viral miRNA called vmiR-5p, which is a miRNA-like viral ncRNA that is expressed by SARS-CoV-2.
Although SARS-CoV-2 does not appear to affect the host miRNAs to an appreciable extent, as shown by the tiny number of small RNA sequences obtained from infected host cells, a potentially valuable observation is that after SARS-CoV-2 infection, the miRNA abundance was increased, whereas two other small ncRNA classes were reduced.
This implies that the virally-induced shut-off of host transcripts does not affect host miRNA production, perhaps because the latter are protected within circulating exosomes.
This findings also points to the potential for using small RNAs to treat COVID-19.
Interestingly among the viral miRNAs, about 5% were 20-nucleotide sequences related to the ORF7a, which is a viral gene that encodes one of the viral accessory proteins and is implicated in innate immunity. The protein is thought to be an antagonist of the type I IFN response.
It has been found that deletions of ORF7a are associated with decreased innate immune evasion. However, these deletions chiefly affect the C-terminal end of the protein, while leaving the N-terminal end, which contains vmiR-5p, intact.
Significantly, this ncRNA is part of a strong hairpin that is highly conserved in coronaviruses. The presence of this hairpin identifies it as a miRNA since hairpin formation occurs during the biogenesis of miRNAs.
Also this vmiR-5p sequence is found in SARS-CoV-2 infection and is positively related to the viral load/genomic RNA.
The study team found that vmiR-5p binds to Ago proteins and may silence host target transcripts. Thus, even though vmiR-5p is expressed at low levels, its association with Ago proteins exploits sequence complementarity that can cause significant target mRNA cleavage while tolerating a small number of mismatched nucleotides.
The study team could not quantitatively assay the effect of vmiR-5p on mRNA transcription within infected cells due to the large-scale suppression of host transcription.
However utilizing synthetic vmiR-5p, the study team found that two predicted target host mRNAs were downregulated.
Interestingly one of these downregulated mRNAs is the Basic Leucine 18 Zipper ATF-Like Transcription Factor 2 (BATF2), which is involved in IFN-gamma signaling. The other mRNA was Heparan Sulfate Proteoglycan 2) HSPG2, which may be implicated in viral superinfection.
Also other possible functions of the vmiR-5p may include the regulation of viral subgenomic or antigenomic RNA. Additional functions of the hairpin itself remain to be elucidated.
Although this miRNA is produced through the host cell’s machinery, it occurs by the processing of a strong hairpin created within the viral ORF7a sequence. This processing requires neither other viral proteins nor Drosha, but may be enhanced by viral genes.
The study team said, “Viruses develop multiple ways to suppress host gene expression, and multiple overlapping mechanisms often evolve. The study findings reveals a new strategy used by SARS-CoV-2, in addition to its known destabilization of host mRNA, its inhibition of translation, splicing, and export.
This RNAi-dependent pathway selectively silences host transcripts, and perhaps even viral transcripts, thus regulating gene expression as necessary for optimal viral replication.”
Host gene variability, virus entry, replication, and antigen presentation
Recent data display that single nucleotide polymorphisms (SNPs) in ACE2 may result in modulation of renin-angiotensin system pathway and associated cardiovascular and pulmonary conditions by altering the angiotensinogen-ACE2 interactions, such as polymorphism Arg514Gly in the African/African-American population. The ACE2 genomic variants may associate with susceptibilities to COVID-19 and cardiovascular complications by altering the AGT-ACE2 pathway (i.e., polymorphism Arg514Gly) [11].
Another mutation Leu584Ala in this gene promotes the entry of SARS-CoV-1 into host cells [69].
Polymorphism His378Arg decreases ACE2 activity, and polymorphism Ser19Pro could distort the most important helix to the S-protein. Other seven missense variants may affect secondary structures (i.e. polymorphisms Gly211Arg; Asp206Gly; Arg219Cys; Arg219His, Lys341Arg, Ile468Val, and Ser547Cys), whereas polymorphism Ile468Val with allele frequency 0.01 is only present in Asia [70].
Human ACE2 variants K26R, S16P, T27A, K31R, H34R, E35K, E37K, D38V, N51S, N64K, K68E, F72V, T921, Q102P, G326E, G352V, D355N, H378R, Q388L, and D509Y are predicted to increase the susceptibility of the individuals carrying these variations to SARS-CoV-2. It has been suggested that the T921I ACE2 variant will favour the improved viral S-protein binding, N90 and T92 ACE2 mutations are critical ACE2 residues that confer protection and are SARS-CoV host modifiers. Variants K31R, E35K, E37K, D38V, N33I, H34R, Q388L, and Y83H in ACE2 were found and are predicted to show a decreased binding to SARS-CoV-2 S-protein, thus protecting individuals corresponding to these genotypes [71].
Based on the genetic analysis, M. Bosso et al. found that the ACE2 polymorphisms associated with hypertension or with the efficacy of ACE1 or angiotensin-receptor blocker could have the potential to alter the binding of SARS-CoV-2 S-protein with ACE2 receptor [72].
The most investigated polymorphism of ACE1 is a genetic deletion/insertion (D/I) polymorphism in intron 16. The D allele is associated with a reduced expression of ACE2. D/I polymorphism has specific patterns of distribution in the European and Asian countries and this variability may be responsible for COVID-19 prevalence in different geographical regions. . About 38% of the prevalence variability can be explained by the relative frequency of the ACE1 D-allele [73]. Patients with the II genotype have lower mortality in comparison with ID and DD genotypes. In contrast, patients with the DD genotype had severe lung infections and high mortality [69].
The ACE I/D polymorphism is associated with many risk factors of COVID-19 severity such as obesity, diabetes, arterial hypertension, heart failure and cancer. The D allele might be associated with risk factor contributions as well as COVID-19 severity and progression [73]. This polymorphism could be important for the complications in COVID-19 patients such as severe lung injury due to the influence on the serum ACE concentration in the whole population. The patients without DD genotype had a lower risk of severe lung injury during COVID-19. Conversely, the presence of the ACE DD allele may be favourable for ACE inhibitors and AT1 receptor blockers therapy [74].
Supporting data were obtained by Gómez J. et al. they found that severe COVID-19 was associated with male gender, hypertension, hypercholesterolemia, and the ACE1 DD genotype [75].
The frequency of the ACE1 II genotype in the European population displays a significant negative correlation with the amount of SARS-CoV-2 cases. Similarly, the ACE1 II genotype is negatively correlated with the number of deaths from SARS-CoV-2 infection [76].
The prevalent polymorphisms in the TMPRSS2 gene, such as Val160Met (rs12329760), are responsible for the risk factor contribution and genetic susceptibility to COVID-19. These risk factors included the high-risk group of male patients and cancer [11]. Val160Met polymorphism influenced any post-translational modifications (e.g., proteolytic cleavage, acetylation, glycosylation, phosphorylation, and sulfation) and decreased the stability of the protein, which prevent the viral entry [77]. The expression quantitative trait loci (eQTLs) variant rs35074065 is linked to the overexpression of TMPRSS2 but with a low expression of the interferon (IFN)-α/β-inducible gene and under-expression of MX1 splicing isoform. This may give rise to enhanced susceptibility to viral infection or a decrease in cellular antiviral response [78, 79].
The involvement of the TMPRSS2 gene variants in the penetration of the virus into cells was described by Torre-Fuentes L. et al. The synonymous variants rs61735792 and rs61735794 showed a significant connection with infection [80]. The presence of nonsynonymous variants TMPRSS11 Arg290Gln (rs353163) and Lys48Arg (rs139010197) may influence the virus penetration into the cell and reduce the level of infection [77].
SARS-CoV-2 has a specific cleavage site between S1 and S2 domains of S protein. This site is critical for the cutting of S protein by the host protease – furin. Furin has some polymorphic variants and the Gly146Ser variant can modulate furin proteolytic activity especially its proprotein convertase activity and its ability to cleave S protein of SARS-CoV-2 [81].
The course and genetic susceptibility in COVID-19 patients may depend on polymorphisms of important regulatory genes – TMPRSS2, CD26 (dipeptidyl peptidase IV), and MX1. Genetic variants of TMPRSS2 (rs112657409, rs11910678, rs77675406, and rs713400) and CD26 (rs13015258) regulated the expression of these important genes during COVID-19. Epigenetic modification at C allele (rs13015258) induces CD26 overexpression which could explain a higher SARS-CoV-2 infected fatality rate in type 2 diabetes [82].
SARS-CoV-2 S protein binds to CD147/BSG receptor. This receptor had increased expression during inflammation and cancer. Inhibition of CD147/BSG can prevent diabetic complications, possibly involving severe lung injury triggers by COVID-19. CD147/BSG has one missense mutation F275V on the I-set domain. Presumably, this mutation might influence the severity of COVID-19 [83,84].
Cathepsin L and cathepsin B (CTSL/B) are determinants of the lysosomal pathway. Genetic changes in the locus of cathepsin L (CTSL1) associated with polymorphism (C-171A) affect the course of hypertension, the C allele associated with higher blood pressure [85]. Vargas et al. suggested a possible connection with SARS-CoV-2 infection of cathepsin gene polymorphisms since the minor allele of one of these polymorphisms (rs41307457) showed a high frequency only in the African population, and similarly, the minor allele rs41312184 was present with a high frequency only in the European population [77].
At the same time, there is insufficient information about the effect of cathepsin gene polymorphisms on the susceptibility to viral infections, their development, and course.
The killer cell lectin-like receptor C2, encoded by the KLRC2 gene, is one of the possible targets in the severity of COVID-19. This receptor had importance for the activation of natural killer cells. A study in a cohort of patients with mild to severe COVID-19 found that genetic variants KLRC2del and HLA-E∗0101 were independent risk factors for severe COVID-19 and may help to identify patients at high risk for severe COVID-19 [86].
The most important process in the recognition of SARS-CoV-2 is an antigen presentation of viral antigen peptides to T and B cells by major histocompatibility complex class I and II molecules (human leukocyte antigens (HLA) in humans).
At present, we have a lack of information about HLA allele polymorphism and arrangements of T and B cell antigen-recognizing receptors (TCRs and BCRs) in the pathogenesis of COVID-19.
In the Asian population, numerous studies have shown the importance of HLA gene polymorphisms for the SARS-CoV-1 induced infection process. The susceptibility and severity of SARS were significantly associated with several HLA class I polymorphisms – HLA-B∗46:01, HLA-B∗07:03, HLA-CW∗08:01 [87, 88, 89] as well as several HLA class II polymorphisms – HLA-DRB4∗01, HLA-DRB1∗12:02 [90, 91]. On the other hand, no evidence of an association between HLA polymorphisms and SARS was found in several studies [92, 93].
The presentation of SARS-CoV-2 antigen peptides to specific CD8+ T cells might be decreased in patients with HLA-A∗02:01 phenotypes in comparison with HLA-A∗11:01 and HLA-A∗24:02 [94, 95]. In COVID-19 patients, HLA class II polymorphisms can crucially influence the antigen presentation.
HLA-DRB1∗08 was associated with low binding of viral antigen peptides and high mortality [96]. Important results were obtained for the identification of HLA-II peptides derived from SARS-CoV-2 spike glycoprotein. Researchers found 526 unique peptides from antigen presenting cells from 9 donors. HLA-II peptides had consensus HLA-II clusters recognized by CD4+ T cells [97].
Other players of SARS-CoV-2 antigen presentation are TCRs and BCRs. Currently there are limited data about the development of T and B cells immunity to SARS-CoV-2. In addition, little is known about the precise regulation of SARS-CoV-2 specific T and B cells differentiation and their persistence.
The key process for the virus-specific differentiation of T and B cells is the clonal V(D)J rearrangements of peripheral TCRs and BCRs. The bioinformatic approach allows comparing TCRs and BCRs from COVID-19 patients and healthy volunteers and to deduce virus-specific TCRs and BCRs.
Recent data showed the high diversity of TCRs in patients with mild COVID-19 and in patients with effective recovery from the disease. Thus, the achievement of huge TCR repertoires may be associated with appropriate immune response and successful outcome in COVID-19.
Likewise T cells, effective B cell response depends on the production of B cell clones with high BCR-antibody activity. Such high BCR-antibody activity might be achieved due to the maturation of somatic hypermutated BCRs during a germinal center reaction. It was shown, that SARS-CoV-2-antibody positive patients had a characteristic pattern of IGHV3 and IGHJ4/6. Unmutated BCRs profile was more characteristics for patients with severe course of COVID-19 [98].
HLA-I allele polymorphism has high importance in the recognition of antigen epitopes in SARS-CoV-2 S glycoprotein for TCRs. For example, HLA-A∗24:02 restricts recognizing by three epitopes S1208-1216, S448-456, and S193-201. Single-cell sequencing of TCR2β has shown that the A24/S448+ CD8+ T cell TCRαβ repertoire depended on a common TCRβ chain motif, while the A24/S1208+ CD8+ TCRαβ repertoire was diverse across COVID-19 patients [99].
In a similar study, the TCRs were compared between healthy volunteers and patients with COVID-19. Single-cell V(D)J analysis revealed 6 VJ pairs (such as TRAV12-2-J27-TRBV7-9-J2-3) significantly increased in patients with COVID-19 [100].
Also, it was shown the monitoring of TCR diversity might be used for the prediction of COVID-19 progression and recovery [101].
The use of TCR repertoire analysis in clinical practice for the prediction of the COVID-19 course is restricted by a large amount of sequencing and data analysis. An open-source software package, tcrdist3, may be used for distance-based analysis and it resolved this problem during SARS-CoV-2 infection [102].
Interferon system
Interferons are part of the body’s antiviral defences and their gene variability influences the susceptibility and severity of COVID-19. It was shown IFN-γ in combination with IFN-β inhibits the replication of SARS-CoV. IFN-γ polymorphic allele +874A was strongly associated with SARS-CoV infection [103].
The single nucleotide polymorphism rs12252-C/C in the IFITM3 gene (which encodes interferon-induced transmembrane protein 3) was detected in patients with COVID-19 and is a risk factor for severe influenza [104]. Y. Zhang et al. also report the relationship between the presence of homozygous polymorphism C of the rs12252 allele in IFITM3 and the development of a more severe course of COVID-19 [105].
The IFITM3 protein showed a potent antiviral capacity to a wide range of viruses, including influenza A viruses, Ebola virus, Marburg virus, SARS-CoV, dengue virus, West Nile virus, Zika virus, and foot-and-mouth disease virus. A strong correlation between the case fatality rate of COVID-19 and the minor allele frequency of the rs6598045 SNP of the IFITM3 gene was identified [106].
Interferon lambda 3 (IFNL3) rs1297860 C/T and INFL4 rs368234815 TT/ΔG gene polymorphisms could affect the ability of the host to modulate viral infection without a clear impact on the outcome of COVID-19 [107].
The large-scale epidemiological data indicate the role of polymorphism A946T (rs1990760) of interferon-induced helicase 1 (IFIH1) in SARS-CoV-2 infection and T allele–carrying individuals may be more resistant to SARS-CoV-2 infection [108].
Proinflammatory cascades
Recently there are limited data about the polymorphisms of several cytokine genes (IFN γ +874A allele, IL12RB1, etc.) and their association with SARS. Other important players in antiviral defence are Toll-like receptor (TLR), which recognise viral components and initiate proinflammatory cascades.
Gene polymorphism of Ticam2, TLR adaptor protein, and its knock out in mice increased susceptibility to SARS-CoV infection [109]. Ticam2 participates in the formation of neutrophil extracellular traps (NETs) [110].
TLRs of the cell surface, especially TLR4, are involved in the recognition of SARS-CoV-2 molecular patterns, causing inflammatory reactions [111].
TLR3 is a receptor for double-strand RNA (dsRNA), and it is associated with the pathogenesis of severe viral infections such as HSV and influenza virus. On the other hand, TLR3 protects against HIV. Replication of many viruses results in the production of dsRNA which induces IFN response [112]. Genetic polymorphisms in other TLRs such as TLR7 and TLR9 led to decreased production of type 1 IFN [113].
TLR3 protein has two polymorphic loci in the extracellular region – N284I and L412F, which modify the receptor response and decrease NF-κB activity. Moreover, human TLR3 has specific residues (His39, His60, His108, His539, and Asn541) which interact with dsRNA, and mutations in these residues might be critical for dsRNA binding and TLR3 signalling [114].
Distribution and prognostic value of Arg753Gln TLR2, Leu412Рhe TLR3, Asp299Gly TLR4 genes polymorphisms in case of grippe have been studied. It has been established that there is an increased risk of grippe development for persons with Asp/Gly TLR4 genotype and TLR2, TLR3, TLR4 mutant genotypes combinations; there is an increased risk of grippe-associated pneumonia for patients with mutant homozygous genotype Phe/Phe TLR3 [115].
In the context of COVID-19 severity interleukin 6 (IL-6) is the most discussed proinflammatory cytokine. IL-6 has clinically important SNP at rs180079 associated with lung diseases, such as chronic obstructive pulmonary disease (COPD), pneumonia. Thus, this IL-6 polymorphism might be relevant to COVID-19 susceptibility and severity [116].
3.4. Oxidative stress/metabolism
Glutathione S-transferase genes T1 (GSTT1, MIM: 600436) and M1 (GSTM1, MIM: 138350) show deletion polymorphism, allele zero. Homozygosity for the null alleles results in a lack of appropriate enzyme activity, which increases the risk of pulmonary fibrosis, one of the most serious complications of COVID-19 disease. Thus, the null genotype GSTT1 can be considered as a predictor of morbidity and mortality from COVID-19 in different geographic regions [117].
The difference in susceptibility to and mortality from COVID-19 might be explained by the presence of vitamin D deficiency due to a different vitamin D metabolism, orchestrated by the DBP polymorphisms of rs7041 and rs4588 [118]. Karcioglu Batur L. described the genetic predisposition to viral infection in vitamin D deficiency and also found a significant correlation between the rs7041 polymorphism, the prevalence of COVID-19, and mortality rates. However, no significant correlation was found between the prevalence (per million) and mortality (per million) at the rs4588 locus [119].
The heme oxygenase-1 (HO-1) is a potential target in COVID-19. Singh D. et al., based on the studies by other authors, suggested a pivotal role of HO-1 induction in inflammation-induced coagulation, observed in COVID-19 patients. The HO-1 genetic polymorphisms, specifically the GT dinucleotide repeat in the promoter region, regulate the inducibility (i.e., transcription) of HO-1 to reactive oxygen species (ROS). Individuals with larger (GT)n repeats have been found to be more susceptible to diseases that involve the endothelium of th
e cardiovascular system, especially in diabetes and obesity. Given the relationship between the presence of GT repeats in the HO-1 promoter region and the severity of disease in various conditions such as acute lung injury, thromboembolism, and diabetes, it is necessary to determine the length of GT repeats in patients with severe COVID-19 [120, 121].
Apoptosis/cell cycle
We observed the lack of data about gene polymorphisms concerned with apoptosis/cell cycle machinery.
Haemocoagulation
Janssen R. proposed a hypothesis that the vitamin K epoxide reductase complex 1 (VKORC1) −1639 A allele had a protective property against inflammation-induced coagulation and decreased the mortality in COVID-19 patients. VKORC1 gene polymorphisms influencing vitamin K metabolism may partially explain the imbalance in morbidity and mortality from COVID-19 in different geographical regions [122].
Karst M et al. have proposed that hyperhomocysteinemia can initiate severe lung injury in COVID-19 patients. This homocysteinemia can be triggered by the presence of the C677T polymorphism of the methylenetetrahydrofolic acid reductase (MTHFR) gene [123].
A separate clinical case showed a positive result for ApoE e3/e4 genes, MTHFR A1298C heterozygous genotype, ACE D/I heterozygous genotype, angiotensinogen M235 heterozygous genotype, and factor XIII (zymogen) Val34Leu heterozygous genotype. A thorough clinical trial has been reported, aiming to validate the hypothesis on the involvement of thrombophilic genetic profiles in the COVID-19 [124].
Rare variants in the gene of plasminogen (PLG), such as Arg261His and Ala494Val, may be recognised as potential markers of inter-individual differences in susceptibility to coronavirus [81].
Recently, the new Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) was described. DC-SIGN bound to different viruses and to S glycoprotein of SARS-CoV. Thus, DC-SIGN can mediate ACE2 independent binding of SARS-CoV-2 to the surface of human cells. Similar properties were characteristic for macrophage galactose lectin (MGL).
DC-SIGN gene polymorphism associated with increased concentration of lactate dehydrogenase (LDH) displaying the high level of systemic inflammation in COVID-19. Other members of the lectin family are also associated with the severity and mortality of COVID-19, for example, mannose-binding lectin. Thus, polymorphic alleles of C-lectin genes might determine the severity of COVID-19 [125].
We summarised the integrating data of possible gene variability as factors of the susceptibility to SARS-CoV-2 and course of COVID-19 with relevant bibliography in Table 1.
reference link :https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8382593/



{kind=link}