









 Extract (6-MSITC) in Healthy Older Adults")
: An In-Depth Exploration into its Thermogenic Role and Social Significance")



A new study by French researchers from the Institut Pasteur-Paris, Université de Paris, Vaccine Research Institute-France and Sorbonne Université-Paris has alarmingly found that the various new emerging SARS-CoV-2 variants display enhanced syncytia formation.
Syncytia is formed by fusion of an infected cells with neighboring cells leading to the formation of multi-nucleate enlarged cells.
This event is induced by surface expression of viral fusion protein that are fusogenic directly at the host cell membrane. Syncytia canonly happen with viruses able to directly fuse at the cellular surface without the need of endocytosis.
Typically, severe COVID-19 is characterized by lung abnormalities, including the presence of syncytial pneumocytes.
The syncytia forming potential of spike variant proteins remain poorly characterized.
The researchers team first assessed Alpha (B.1.1.7) and Beta (B.1.351) spread and fusion in cell cultures, compared to the ancestral D614G strain. Alpha and Beta replicated similarly to D614G strain in Vero, Caco-2, Calu-3 and primary airway cells.
However, Alpha and Beta formed larger and more numerous syncytia. Variant spike proteins displayed higher ACE2 affinity compared to D614G.
Alpha, Beta and D614G fusion was similarly inhibited by interferon induced transmembrane proteins (IFITMs). Individual mutations present in Alpha and Beta spikes modified fusogenicity, binding to ACE2 or recognition by monoclonal antibodies.
The study findings were published in the peer reviewed EMBO Journal.
reference link :https://www.embopress.org/doi/abs/10.15252/embj.2021108944
Syncytia are evolutionarily conserved cellular structures form by the multiple cell fusions of uninuclear cells. In mammals, the best example of physiological syncytia is muscle fibers, which contain thousands of fused muscle cells to allows their rapid coordinated contraction [7].
It is also important in the decidualization process during embryo implantation [8]. Syncytia can also be induced by certain types of infections by viruses, such as human immunodeficiency virus, respiratory syncytial virus, and herpes simplex virus [9].
It could be envisioned that virus-induced cell fusion facilitates the transfer of viral genomes to the neighboring cells. However, the viral and cellular mechanisms regulating the formation of syncytia during SARS-CoV-2 infection remains largely elusive.
While examining the histopathologic lung sections from patients died from COVID-19, the Giacca group and the Sun group observed the prevailing existence of atypical cells containing 2-20 nuclei.
The identities of these syncytia were later confirmed by their expression of pneumocyte specific makers. In vitro co-culture assay showed that monkey kidney epithelial cell line, Vero cells (ACE2+), upon expressing the SARS-CoV-2 spike protein, could form homologous syncytia or fuse with other cell lines as long as the ACE2 receptor was present.
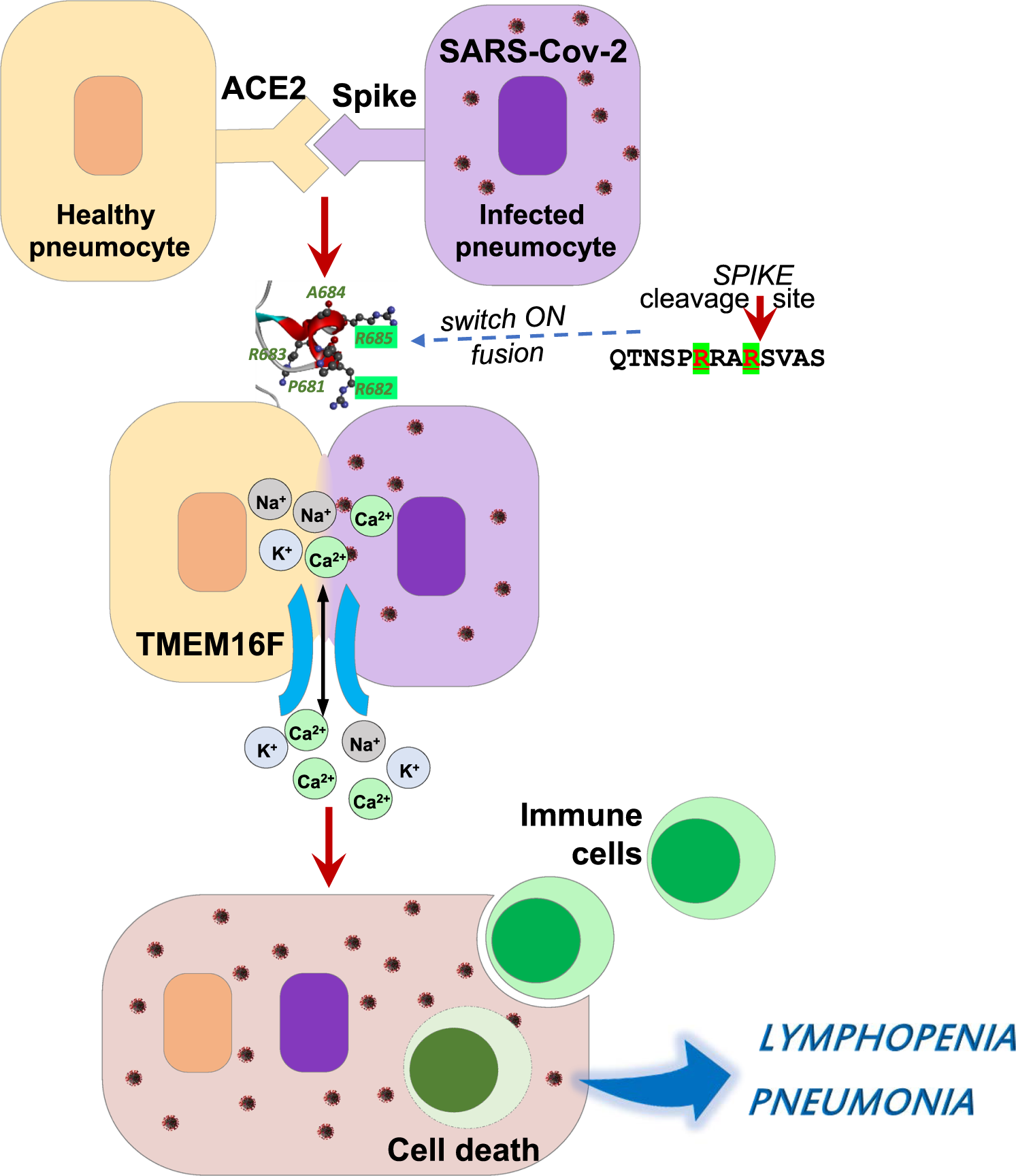
Interestingly, when Vero cells were transfected with Spike protein from SARS-CoV-1, no formation of syncytia was observed. Therefore, the key element responsible for SARS-CoV-2-mediated syncytia is absent in the spike protein of SARS-CoV-1. Driven by this hypothesis, Sun and et al. compared the spike protein from SARS-CoV-2 and SARS-CoV1 and found that there is a four amino acids (PRRA) insertion before the S1/S2 cleavage site in the SARS-CoV-2 spike protein.
The truncated mutation of SARS-CoV-2 spike protein with “PRRA” deletion lose its abilities to fuse cells. Consistently, spike protein from SARS-CoV1 effectively induced syncytia once the “PRRA” sequence was inserted before the S1/S2 cleavage site of the SARS-CoV-1 genome.
Furthermore, the Sun group demonstrated that a bi-arginine motif containing R682 and R685 dictates syncytium formation by constructing single or combine mutations in the “PPRA” insertion. Fig. 1.
Fig. 1: The SARS-CoV-2 spike protein and cellular TMEM16 ion channel collaboratively mediated the formation of syncytia in COVID-19 infections.
The new data obtained by Sun et al. provide critical information for understanding syncytia through deciphering the structure basis required for SARS-CoV-2 spike protein-mediated cell fusion, while Giacca et al. focused on the cellular mechanism and therapeutic potential of syncytia during SARS-Cov-2 infection.
In this regard, Giacca and colleagues screened 3049 FDA/EMA-approved drugs using SARS-CoV-2 spike protein expressing Vero cell-based in vitro cell fusion system to search for drugs that block syncytia.
Interestingly, drugs that suppressed cell fusion are all capable of regulating intracellular Ca2+ levels. Among the syncytia blocking drugs, niclosamide, an oral anti-helminthic agent, was found to be effective at a very low dose (IC50 = 0.34 μM) and could prevent cell from virus-induced cell death.
Niclosamide is a potent antagonist of Ca2+-activated TMEM16/anoctamin family of chloride channels [10]. TMEM16F was also dramatically increased in Vero cells upon spike protein expression. When TMEM16F expression was disturbed, the syncytia formation in spike-expression cells were diminished. Therefore, TMEM16F activation is the signal responsible for triggering syncytia.
These two elegant studies collectively revealed a new concept of syncytia formation and its roles in SARS-CoV-2 infections, which can be briefly précised as follows. The SARS-CoV-2 infections induce the surface expression of the spike glycoprotein.
The interaction of the spike protein with the ACE2 receptor of the neighboring cells then activate TMEM16F, and trigger the unshealthe of the profusion S2 fragment of the spike protein in a bi-arginine motif dependent manner, which eventually leads to the membrane fusion and syncytia formation. However, there are still many questions that remain to be elucidated.
One of which is whether the bi-arginine motif is required for the activation of TMEM16F. Another is the impact of syncytia formation on SARS-CoV-2 infections in vivo. Sun et al. found that a type of CD45 positive cell structure presents in the syncytia of the COVID-19 patients.
This could be a cell-in-cell structure. When human peripheral blood mononuclear cells were co-cultured with SARS-CoV-2 spike protein-induced syncytia, they could be engulfed by and die inside the syncytia, thus providing a possible explanation for lymphopenia in SARS-CoV-2 infections [11].
It can be highly suspected that syncytia are deleterious for COVID-19 patients since syncytia were observed only in the severe stages of the diseases and syncytia may induce lymphopenia. Despite the observation of multinucleate pneumocytes in autopsy, it is still not known whether such syncytia play a critical role in the pathogenesis of CRDs of severe COVID-19 patients. Recently, an antidepressant drug, fluvoxamine, was shown to lower the likelihood of clinical deterioration of severe COVID-19 patients in a randomized clinical trial [12].
Interestingly, fluvoxamine could facilitate TMEMF activation and phosphatidylserine exposure [13]. It is imperative to examine whether fluvoxamine affects syncytia formation. It is also worthy to evaluate the whether the combine uses of anti-syncytia drugs with other COVID-19 targets would yield better clinical outcomes [14, 15].
Overall, these two papers provide critical information for the understanding of how syncytia occurred during SARS-CoV-2 infections at the virus structure and cellular signaling points of views and open up a new revenue in COVID-19 studies. It is anticipated that these novel findings may provide information for developing new strategies to combat the current pandemic.
reference link : https://www.nature.com/articles/s41418-021-00795-y
Discussion – reference link :https://www.embopress.org/doi/abs/10.15252/embj.2021108944
The replication and cytopathic effects of SARS-CoV-2 variants is under intense scrutiny, with contrasting results in the literature (Frampton et al., 2021; Hou et al, 2020; Leung et al, 2021; Liu et al, 2021b; Touret et al., 2021). For instance, there was no major difference in the replication kinetics of Alpha and D614G strains in some reports (Thorne et al, 2021; Touret et al., 2021), whereas others suggested that Alpha may outcompete D614G in a co-infection assay (Touret et al., 2021). Other studies proposed that the N501Y mutation may provide a replication advantage, whereas others suggested that N501Y is deleterious (Frampton et al., 2021; Hou et al., 2020; Leung et al., 2021; Liu et al., 2021b). These discrepant results may be due to the use of different experimental systems, viral strains, multiplicities of infection and cell types.
Here, we show that Alpha and Beta variants replicate to the same extent as the early D614G strain in different human cell lines and primary airway cells. Moreover, Alpha and Beta induced more cell-cell fusion than D614G. Increased fusion was observed in U2OS-ACE2 cells and in naturally permissive Vero cells. In agreement with infection data, transfection of Alpha and Beta S proteins, in the absence of any other viral factors, produced significantly more syncytia than D614G, which in turn, fused more than the Wuhan S.
Comparative video microscopy analysis revealed that Alpha S fused the most rapidly, followed by Beta, D614G, and finally Wuhan. Thus, Alpha and Beta variants display enhanced S-mediated syncytia formation. One limitation of our study resides in the fact that were unable to look at surface expression of the variant S proteins in Vero and Caco2 without losing the large S protein positive syncytia.
We thus used the non-fusogenic 293T cells to control for surface expression. We further show that S-expressing 293T cells fuse with Vero cells in donor/acceptor experiments. The experiments confirmed the enhanced fusogenicity of the variants in cells with similar levels of S protein at their surface.
We further show that Alpha and Beta remain sensitive to restriction by IFN-β1. The fusion mediated by their respective S proteins is inhibited by IFITMs. This extends previous results by us and others demonstrating that ancestral Wuhan S is effectively inhibited by this family of restriction factors (Buchrieser et al., 2020; Shi et al., 2021).
It has been recently reported in a pre-print that Alpha may lead to lower levels of IFN-β1 production by infected Calu-3 cells and may be less sensitive to IFN-β pre-treatment, when compared to first wave viral isolates (Thorne et al., 2021).
We did not detect here differences of IFN-β1 sensitivity between the variants in Vero and U2OS- ACE2 cells. Again, these discrepant results may reflect inherent differences between Calu-3, Vero and U2OS-ACE2 cells, or the use of different viral isolates.
We then characterized the contribution of the individual mutations present in Alpha and Beta S proteins to their respective fusogenicity. The highly fusogenic Alpha S consists of more mutations that robustly increase fusion (P681H and D1118H) than mutations that decrease fusion (∆69/70).
In contrast, the Beta variant is comprised of several restrictive mutations (∆242-244, K417N, and E484K) and only one mutation that modestly increased fusion (D215G). The strongest increase in fusion was elicited by the P681H mutation at the S1/S2 border. This mutation likely facilitates proteolytic cleavage of S and thus promotes S mediate cell-cell fusion. Indeed, the analogous P681R mutation present in B.1.617.2 and B.1.617.3 variants increases S1/S2 cleavage and facilitates syncytia formation (Ferreira et al, 2021; Jiang et al, 2020).
Of note, another report with indirect assessment of variant S fusogenicity suggested a mild decrease or no difference in cell-cell fusion of Alpha and Beta relative to Wuhan S (Hoffmann et al, 2021). These previous experiments were performed in 293T cells at a late time-points (24 hours post-transfection), which may preclude detection of an accelerated fusion triggered by the variants.
We show that the binding of variant S to soluble ACE2 paralleled their fusogenicity. Alpha bound the most efficiently to ACE2, followed by Beta, D614G and finally Wuhan. However, the ACE2 affinity of S proteins carrying individual mutations did not exactly correlate to fusogenicity. For instance, the N501Y and D614G mutations drastically increased ACE2 affinity, but only D614G enhanced fusogenicity.
The K417N substitution, and to a lesser degree ∆242-244, had a lower affinity to ACE2 and also restricted cell-cell fusion. The E484K mutation significantly restricts fusion, but mildly increases ACE2 affinity. This suggests that on the level of individual S mutations, the relationship between ACE2 affinity and increased fusogenicity is not always linear. Variant mutations may also confer advantages in an ACE2 independent manner.
Indeed, recent work has suggested that the E484 mutation may facilitate viral entry into H522 lung cells, requiring surface heparan sulfates rather than ACE2 (Puray-Chavez et al, 2021). It would be of future interest to examine the syncytia formation potential of the variant mutations in other cell types.
We selected a panel of 4 mAbs that displayed different profiles of binding to Alpha, Beta, D614G and Wuhan S proteins. The mAb10 targeting the S2 domain recognized all variants and was used as a positive control. Wuhan and D614G were recognized by the three other antibodies, targeting either the NTD or RBD. Alpha lost recognition by the anti-NTD mAb71, whereas Beta was neither recognized by mab71 nor by the two anti-RBD antibodies mAb48 and mAb 98. Upon examining the potential of S proteins carrying individual mutations to bind to human monoclonal antibodies, we found that the ones that restrict (∆242-244, K417N) or have no effect on fusogenicity (∆Y144) are also not recognized by some mAbs.
This suggests that variant S proteins have undergone evolutionary trade off in some circumstances; selecting for mutations that provide antibody escape at the detriment of fusogenicity. In accordance with our findings, deep sequence binding analysis and in vitro evolution studies suggest the N501Y mutation increases affinity to ACE2 without disturbing antibody neutralization (Liu et al, 2021a; Starr et al., 2021; Zahradník et al, 2021).
The E484K and K417N RBD mutations in the Beta variant may also increase ACE2 affinity, particularly when in conjunction with N501Y (Zahradník et al., 2021) (Nelson et al, 2021). However, the resulting conformational change of the S protein RBD may also decrease sensitivity to neutralizing antibodies (Nelson et al., 2021). Future work assessing the structural and conformational changes in the S protein elicited by a combination of individual mutations or deletions may further help elucidate the increased fusogenicity and antibody escape potential of the variants.
While we had previously shown that the interaction between the S protein on the plasma membrane with the ACE2 receptor on neighboring cells is sufficient to induce syncytia formation, there is compelling evidence of the importance of the TMPRSS2 protease in S activation (Buchrieser et al., 2020; Dittmar et al, 2021; Koch et al, 2021; Ou et al, 2021). We found that the S protein of the novel variants induced more syncytia formation than the D614G and Wuhan S proteins in human Caco2 cells which express endogenous ACE2 and TMPRSS2. However, we did not detect any major
differences in the processing of the variant S proteins by TMPRSS2. It will be worth further characterizing how the fusogenicity of variant associated mutations are influenced by other cellular proteases like furin.
The presence of infected syncytial pneumocytes was documented in the lungs of patients with severe COVID-19 (Bussani et al., 2020; Tian et al, 2020; Xu et al, 2020). Syncytia formation may contribute to SARS-CoV-2 replication and spread, immune evasion and tissue damage. A report using reconstituted bronchial epithelia found that viral infection results in the formation and release of infected syncytia that contribute to the infectious dose (Beucher et al., 2021).
The neutralizing antibody response to SARS-CoV-2 infection has divergent effect on cell-cell fusion, with some antibodies restricting S mediated fusion, while other increase syncytia formation (Asarnow et al, 2021). Cell-to-cell spread of virus may be less sensitive to neutralization by monoclonal antibodies and convalescent plasma than cell-free virus (Jackson et al, 2021). It is thus possible that infected syncytial cells facilitate viral spread. Within this context, it is necessary to better understand the fusogenic potential of the SARS-CoV-2 variants that have arisen and will continue to emerge.
We have characterized here the replication, fusogenicity, ACE2 binding and antibody recognition of Alpha and Beta variants and the role of their S-associated mutations. Despite the insights we provide into the S-mediated fusogenicity of the variants, we did not address the conformational changes that the mutations individually or in combination may elicit.
We further show that Alpha, Beta and Delta S proteins more efficiently bind to ACE2 and are more fusogenic than D614G. Which virological and immunological features of the Delta variant explain its higher estimated transmissibility rate than Alpha and other variants at the population level remains an outstanding question.




{kind=link}